
Abstract
This narrative review explores the mechanisms underlying resistance to cyclin-dependent kinase 4/6 inhibitors (CDK4/6i) in hormone receptor (HR)-positive metastatic breast cancer (MBC), a critical challenge in contemporary oncology. Despite the proven efficacy of CDK4/6i in improving clinical outcomes, both intrinsic and acquired resistance remain substantial challenges. We discuss clinical data that underscore pivotal molecular alterations associated with resistance, including mutations in the Retinoblastoma gene (RB1), germline variants, aberrations in the PIK3CA gene that activate the phosphatidylinositol 3-kinase/protein kinase B (AKT)/mammalian target of rapamycin signaling cascade, and modifications in fibroblast growth factor receptor signaling. Additional resistance mechanisms—such as the loss of the FAT1 tumor suppressor gene and the dysregulation of cell cycle regulators like cyclin E and CDK2—are also explored. The role of circulating tumor DNA analysis in tracking genomic changes during therapy is also considered. Furthermore, the review assesses emerging therapeutic strategies, particularly combination therapies that target alternative pathways to counteract resistance mechanisms. By synthesizing current evidence and providing actionable insights, this review aims to enhance our understanding of endocrine resistance mechanisms among clinical oncologists and gives them some future perspectives to expand strategies to overcome this challenge.
Plain language summary
This review focuses on why some hormone receptor-positive metastatic breast cancers do not respond to CDK4/6 inhibitors, which are effective treatments. It examines both primary and developed resistance to these drugs. Key genetic changes linked to resistance include mutations in the retinoblastoma gene (RB1), variations in the PIK3CA gene activating the PI3K/AKT/mTOR pathway, and changes in fibroblast growth factor receptor (FGFR) signaling. The article also discusses other resistance factors, such as the loss of the FAT1 tumor suppressor gene and disruptions in cell cycle regulators like cyclin E and CDK2. Additionally, the review evaluates how circulating tumor DNA (ctDNA) can help monitor genetic changes during treatment. It highlights new therapeutic strategies, including combination therapies that target different pathways to combat resistance. By compiling and interpreting current research, this review aims to deepen the understanding of how hormone-related resistance mechanisms work. It ultimately seeks to provide oncologists with insights and future directions to improve treatment strategies against resistance in this challenging area of breast cancer care.
Keywords
Introduction
In recent decades, therapeutic advancements have significantly improved survival rates for women diagnosed with hormone receptor-positive (HR+) breast cancer. Analysis of population-based databases indicates that between 2015 and 2019, there was a remarkable 22% reduction in overall mortality and a 27% decrease in breast cancer-specific mortality compared to those diagnosed in the early 1990s. 1 These enhanced outcomes can largely be attributed to substantial progress in endocrine and targeted treatment strategies, particularly the introduction of cyclin-dependent kinase 4/6 inhibitors (CDK4/6i), which have transformed patient management and prognosis.2,3
Specific patient groups show de novo (intrinsic or primary) resistance to these agents, while those who respond often encounter acquired (or secondary) resistance and disease progression. Within the continuum of resistance mechanisms, distinguishing intrinsic from acquired resistance is complex. Intrinsic resistance may arise from pre-existing factors such as CDK overexpression or retinoblastoma (Rb) gene loss, interactions with BRCA2 mutations, and the role of the intrinsic non-luminal subtype. In contrast, acquired resistance typically develops during treatment due to genetic mutations or alternative signaling pathways. Despite advances in understanding CDK4/6i resistance, translational analyses from pivotal trials have failed to identify applicable biomarkers in clinical practice. The lack of reliable biomarkers for identifying potential responders and early resistance mechanisms to CDK inhibitors poses significant challenges for implementing precision medicine in advanced hormone HR+ breast cancer.4–6
This review comprehensively examines the latest research and emerging perspectives on resistance to CDK4/6i in HR+ metastatic breast cancer (MBC). By synthesizing existing evidence regarding resistance mechanisms, this article aims to provide clinical oncologists with actionable insights to refine current treatment practices and guide future therapeutic developments in this crucial area of oncology.
The evolution of CDK4/6i development
CDKs are essential for regulating cancer cell proliferation, as they disrupt cell cycle control mechanisms, expressly the G1 to S phase transition governed by the cyclin D-CDK4/6-INK4-RB axis. Upregulation of cyclin D via mitogenic pathways, including steroid hormones and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling, leads to active cyclin D-CDK4/6 complexes. 7 These complexes phosphorylate Retinoblastoma gene 1 (RB1), causing the release of E2F transcription factors that drive genes necessary for DNA replication and cell cycle progression. By targeting and disrupting the dysregulated CDK4/6-Rb-E2F pathway, CDK inhibitors effectively halt cell proliferation, especially in HR+ breast cancer.8,9 The development of these inhibitors has evolved from early nonselective agents like flavopiridol and roscovitine, which caused significant off-target effects, to more selective CDK4/6i designed for enhanced specificity and improved therapeutic windows.8,10
The advent of CDK4/6i has notably transformed the treatment of HR+ MBC. Pivotal first-line (1L) clinical trials have reported significant improvements in survival, with a decreased risk of disease progression or death since including CDK4/6i in standard treatment protocols post-2015.11–13 Survival benefits have been extensively validated by secondary studies, including systematic reviews and meta-analyses. 14 These developments are central to understanding CDKs, which are essential for cancer cell proliferation. 15 While earlier generations of CDK4/6i encountered challenges with non-selectivity and toxicity, third-generation selective inhibitors have demonstrated effective targeting of specific CDKs, critical for cell cycle progression. 16
The adverse event profiles of the three widely utilized CDK4/6i (palbociclib, ribociclib, and abemaciclib) differ in terms of clinical implications for breast cancer treatment. 17 Palbociclib and ribociclib are primarily associated with hematological toxicities such as neutropenia, leukopenia, anemia, and thrombocytopenia, with neutropenia being the most frequent and requiring monitoring despite its reversible nature. Abemaciclib, on the other hand, is predominantly linked to gastrointestinal side effects, peculiarly diarrhea, affecting up to 92% of patients and impacting the quality of life. 18 Ribociclib presents higher risks of hepatic toxicity, respiratory complications, and QT interval prolongation, principally in metastatic cases. Although less common, these agents can also result in cutaneous and cardiovascular toxicities, including dermatological reactions, alopecia, arrhythmias, and thromboembolic events. 19 Some factors can be associated with a higher risk for toxicity, as advanced age and low body mass index were linked to a greater incidence of neutropenia in an Italian real-world study. 20 Understanding these distinct toxicity profiles is essential for clinicians to optimize treatment and supportive care while minimizing adverse effects on patients. 21
Mechanisms of resistance and emerging therapeutic opportunities in clinical practice
Figure 1 illustrates the primary mechanisms of resistance to CDK4/6i, along with the suggested therapeutic agents to overcome these challenges.

Main mechanisms of resistance to CDK4/6i and proposed therapeutic agents.
Molecular alterations in RB1
RB1 serves as a pivotal tumor suppressor, with its product, the retinoblastoma protein (pRB), being the main target of the Cyclin D-CDK4/6 complex. The integrity of RB1 is essential for the effectiveness of CDK4/6i; however, alterations in this gene can indicate mechanisms of intrinsic or acquired resistance. 22 Studies in translational and molecular epidemiology have demonstrated that baseline mutations in RB1 are uncommon, with frequencies reported between 0 and 5% in circulating tumor DNA (ctDNA) analyses.23,24 Tumors with RB1 alterations have been shown to experience significantly reduced progression-free survival (PFS) benefits when treated with CDK4/6i. 25
Recent research emphasizes the role of specific RB1 mutations, expressly “double-hit” events, in conferring endocrine resistance to CDK4/6i in breast cancer. A “double-hit” consists of the simultaneous inactivation of both alleles of the RB1 gene, which may occur through complete gene deletions, point mutations, or loss of heterozygosity (LOH), resulting in a total loss of RB1 function. 26 This genetic alteration exacerbates genomic instability and enhances tumor survival mechanisms, thereby contributing to the aggressive nature of these malignancies. Advanced techniques, such as liquid biopsies for ctDNA analysis, allow real-time monitoring of these alterations and provide insights into tumor evolution and RB1 status. 27 Following progression on CDK4/6i, patients with RB1-deficient tumors may benefit from alternative treatment strategies, such as PARP inhibitors or agents targeting downstream effectors of RB1. 28 Thus, comprehensive genetic profiling, including a continuous assessment of RB1 mutations and double-hit events, is crucial for stratifying treatment options and optimizing outcomes in breast cancer management.
In the PALOMA-3 trial, it was found that emerging RB1 mutations were identified in only 4.7% of patients in the palbociclib plus fulvestrant arm. 29 These alterations were not identified at baseline, highlighting the acquisition or selection of RB1 aberrations under pressure from CDK4/6i treatment. Further supporting this, Li et al. 25 described that patients with RB1 loss experienced considerably worse outcomes, with a median PFS of 3.6 months compared to 10.1 months for those with intact RB1 when treated with CDK4/6i and endocrine therapy (ET). Similarly, patients showing inactivating RB1 mutations or deletions in ctDNA from the MONALEESA trials derived lower survival benefit from ribociclib, with a median PFS of 3.8 compared to 9.2 months in the RB1 wild-type population. 30
Findings from the YoungPearl study, which involved premenopausal patients, also linked RB1 loss (observed in 4% of participants) to shorter PFS intervals in those receiving palbociclib and ET. 31 Palafox et al. 26 identified heterozygous RB1 loss as a biomarker of acquired resistance and poorer clinical outcomes. Their study revealed worse disease-free survival, overall survival (OS), and duration of treatment in patients with RB1 loss. A notable association was also established between double-hit alterations (mutation and heterozygous deletion) and previous exposure to CDK4/6i, suggesting that tumors with heterozygous RB1 loss are likely to acquire additional mutations, thereby contributing to resistance.
Dysregulation of cell cycle machinery
Dysregulation of cell cycle machinery is a hallmark of cancer, leading to unchecked cellular proliferation. 32 Tumors exhibiting overexpression or copy number amplification of cyclin D or CDK4/6, alongside inactivation of CDKN2A genes (key regulators of CDK4/6 activity), may show heightened sensitivity to CDK4/6i. Furthermore, downstream alterations, such as RB1 inactivation or increased activity of E2F and cyclin E-CDK2 complexes, can promote cell cycle progression independently of CDK4/6 function. 33 Despite these findings, the only clinically validated biomarker for therapy with CDK4/6i still remains estrogen receptor (ER) positivity. 34
HR+ tumors are often linked to cyclin D1 amplification and/or CDKN2A inactivation, suggesting that proliferation in these tumor cells is predominantly driven by hormonal factors through cyclin D1-CDK4/6 activity. 35 Compelling evidence also indicates the adaptive activation of CDK2 in response to CDK4/6 inhibition. Single-cell analysis of CDK2 activity in triple-negative breast cancer (TNBC) cell lines—distinguished by sensitive (luminal) and resistant (basal) phenotypes—demonstrated that CDK2 can be activated independently of CDK4/6, facilitating the transition from the G1 to the S phase of the cell cycle.36,37
Moreover, investigations have shown that cyclin E1 gene (CCNE1) amplification or elevated expression can impact the efficacy of CDK4/6i, positioning cyclin E1 as a potential intrinsic biomarker of resistance. 37 In the PALOMA-3 trial, palbociclib demonstrated lower efficacy in patients with high CCNE1 RNA expression at baseline. Conversely, cyclin E1 levels had no effect on clinical outcomes for patients treated with placebo/fulvestrant. 38 Despite this association, CCNE1 remains absent from clinical biomarker panels due to several challenges, including the lack of standardized assays for measuring its expression and the regulatory requirement for extensive validation through large clinical trials. Consequently, clinicians may encounter barriers to adopting CCNE1 as a biomarker without robust evidence of its predictive value across diverse populations and treatment contexts. 39
Additionally, related translational studies from the PALOMA-2/3 and MONALEESA-2 trials found no significant correlation between CCNE1 expression and survival outcomes.40,41 While extensive preclinical and translational research has provided valuable insights into the role of cyclin E1, the integration of biomarkers associated with cell cycle control into clinical practice remains primarily experimental in breast cancer. 42 Additional endorsement is required before these biomarkers can be routinely integrated into clinical decision-making, potentially enhancing the personalization of treatment strategies and improving prognosis.
ACK1 activation in breast cancer triggers epigenetic reprogramming through the phosphorylation of histone H4 at tyrosine 88 (Tyr88), which promotes the transcription of critical cell cycle genes, including CCNB1, CCNB2, and CDC20. This increased expression facilitates the G2/M transition, enabling cancer cells to proliferate even in the presence of CDK4/6i like palbociclib, thereby contributing to resistance against these targeted therapies. 43 Preclinical studies suggest that inhibiting ACK1 with the small molecule (R)-9b disrupts this epigenetic modulation, leading to reduced transcription of cell cycle genes and resensitizing breast tumor cells to CDK4/6i. These findings underscore the pivotal role of ACK1 in mediating drug resistance and indicate that targeting ACK1 could offer a novel therapeutic strategy to enhance the efficacy of CDK4/6i in cases of resistant breast cancer. 44
Alterations in fibroblast growth factor receptor
Alterations in the fibroblast growth factor receptor (FGFR) pathway contribute to resistance to CDK inhibitors in endocrine-resistant breast cancer, as fibroblast growth factors (FGFs) signal through this receptor, regulating cellular proliferation and survival. Mutations in FGFR2, such as N550K and M538I, have been linked to resistance to both fulvestrant and palbociclib in HR+ cells. In cells resistant to ET, FGFR1 overexpression leads to heightened activation of the PI3K/AKT and RAS/MEK/ERK signaling pathways in response to FGF2, sometimes resulting in ligand-independent signaling. 45
Recent analyses of ctDNA from patients treated with palbociclib and ET identified FGFR alterations in 14 of 34 samples, primarily FGFR1 amplifications. 46 In the MONALEESA-2 trial, 5% of patients exhibited FGFR1 changes, which correlated with reduced PFS compared to wild-type patients (10.6 vs 24.8 months, p = 0.075). 45 However, the efficacy of ribociclib was maintained regardless of FGFR1 status.47,48 The potential for combining FGFR inhibitors with CDK4/6i and ET is actively being explored. A phase II trial demonstrated that the combination of fulvestrant with the FGFR inhibitor dovitinib improved median PFS (10.9 vs 5.5 months for placebo). 6
Ribociclib and palbociclib may differ in their binding affinities and pharmacodynamic effects, impacting their efficacy against FGFR1-altered tumors. FGFR1 amplifications could lead to compensatory activation of alternative signaling pathways, making tumors more resistant to palbociclib by enhancing downstream signaling. 46 In contrast, ribociclib may provide more effective CDK4/6 inhibition in these settings, disrupting both the cell cycle and compensatory pathways, potentially leading to improved anti-tumor effects. In addition, ribociclib might influence the tumor immune microenvironment differently than palbociclib, which could further enhance therapeutic efficacy.47,48 Variations in pharmacokinetics and FGFR pathway dynamics could also result in differing clinical outcomes, with ribociclib offering more sustained inhibition of cell proliferation in tumors with high FGFR signaling despite possible resistance mechanisms.
Loss of FAT1 tumor suppressor, Hippo pathway inactivation, and CDK6 overexpression
Tumor suppressor genes, such as FAT1, are crucial for regulating cellular processes including growth, differentiation, and apoptosis (Figure 1). 25 The loss of function mutations affecting FAT1 can disrupt the mechanisms that typically restrain Cell proliferation, leading to unchecked cancer cell growth, particularly in HR+ breast cancers. In these cases, alterations in tumor suppressor genes are markedly supposed to affect treatment outcomes and patient prognosis. 49
One pivotal pathway impacted by the loss of FAT1 is the Hippo signaling pathway, which is essential for maintaining tissue homeostasis and regulating cellular proliferation. 50 The Hippo pathway inhibits the activity of transcriptional co-activators YAP and TAZ; when these factors are activated, they promote the expression of genes involved in cell cycle progression and cell survival. When functional tumor suppressors like FAT1 are absent, the Hippo pathway is often suppressed, resulting in the nuclear localization of YAP/TAZ and the subsequent upregulation of oncogenic factors, including CDK6. 51 This dysregulation not only promotes cellular proliferation but also contributes to resistance against CDK4/6i. 49
Previous findings indicate that FAT1 mutations are found in only 2%–6% of HR+/HER2− breast cancers. Patients with FAT1 loss demonstrate intrinsic resistance to CDK4/6i and experience reduced PFS when these agents are used alongside ET. 25 The infrequency of FAT1 mutations limits the clinical utility of routine screening for FAT1 and Hippo pathway alterations in oncology. Consequently, the challenges and costs associated with detection may not justify their inclusion in standard practice. Furthermore, the limited therapeutic options for targeting downstream effectors like CDK6 or YAP/TAZ highlight the need for more effective treatment strategies.
Alterations in the TP53 pathway (TP53 loss of function mutations, MDM2 amplifications)
The significance of TP53 mutations in breast cancer has garnered considerable attention due to their association with treatment resistance, especially in relation to CDK4/6i. As a critical tumor suppressor gene, TP53 is frequently altered across various malignancies, including HR+ breast cancer. Research indicates that loss-of-function mutations in TP53 are prevalent among patients who exhibit poor long-term responses to CDK4/6i therapy. These mutations disrupt the essential regulatory functions of tumor protein p53, which include cell cycle regulation, DNA damage response, and apoptosis, thereby promoting tumor cell survival and proliferation despite therapeutic interventions. Approximately 30%–40% of HR+ breast cancer cases harbor TP53 mutations, highlighting the urgent need for understanding the clinical implications and potential targeted therapeutic strategies in this patient population.52,53
The mechanisms through which TP53 mutations confer resistance to CDK4/6i are complex. In normal cells, functional p53 allows CDK4/6i to induce cell cycle arrest by inhibiting the phosphorylation of pRB, thus preventing cell cycle progression. In contrast, in cells carrying TP53 mutations, this regulatory pathway is compromised, allowing mutant p53 to cause aberrant re-entry into the cell cycle and escape from quiescence. This enables continuous proliferation of cancer cells, even in the presence of CDK4/6i.54,55 Kudo et al. 56 evaluated 467 patients with HR+ MBC and found that 129 patients (27.6%) harbored loss-of-function variants in TP53, while 30 (6.4%) exhibited MDM2 amplifications prior to initiating CDK4/6i therapy, alterations that were associated with reduced PFS.
In response to the challenges posed by TP53-mediated resistance, there is growing interest in developing alternative therapeutic options for patients with HR+ breast cancer-bearing TP53 mutations. One promising approach involves the selective inhibition of CDK2, which has demonstrated the potential to counteract the effects of p53 loss and restore cell cycle control. By targeting CDK2, it may be possible to enhance the therapeutic response in tumors with TP53 mutations, potentially improving clinical outcomes. Furthermore, integrating genetic profiling into clinical practice could facilitate the identification of patients who are likely to benefit from such targeted therapies, paving the way for more personalized treatment strategies.57,58
Preclinical data have demonstrated the potential of MDM2 antagonists as an effective strategy to combat intrinsic resistance to CDK4/6i in breast cancer therapy.59,60 These antagonists stabilize the tumor suppressor p53, which subsequently increases the expression of the cyclin-dependent kinase inhibitor p21. This mechanism is beneficial to a large extent for tumors retaining wild-type TP53, suggesting improved therapeutic outcomes. Certain breast cancer cases exhibit co-amplifications of MDM2 and CDK4, a condition associated with resistance to CDK4/6i. 61 Mutations in TP53 are frequently linked to poor treatment responses, emphasizing its critical role in therapeutic efficacy. The combination of MDM2 antagonists with CDK4/6i has been shown to enhance anti-tumor activity and prolong cell cycle arrest in tumors expressing functional p53. 62
Aurora kinase upregulation
Aurora kinase A (AURKA) has emerged as a critical factor mediating resistance to CDK4/6i in HR+ MBC. AURKA is a serine/threonine kinase essential for cell cycle regulation, in particular during mitosis. Alterations in AURKA, such as gene amplification and overexpression, are prevalent in a substantial subset of patients who display resistance to CDK4/6i therapies. Research indicates that AURKA amplifications are found in approximately 26.8% of tumor samples from resistant patients, whereas these alterations are mainly absent in samples from those who respond to treatment. 23 This finding implies that AURKA alterations may facilitate resistance mechanisms, enabling cancer cells to evade the growth-inhibitory effects of CDK4/6i, thus promoting tumor progression and poorer survival outcomes.
The presence of AURKA alterations has been associated with unfavorable clinical outcomes in individuals receiving CDK4/6i. Specifically, patients with AURKA amplification tend to exhibit shorter PFS compared to those without such alterations. Furthermore, the role of AURKA in mediating resistance is highlighted by its participation in various signaling pathways that enhance cell proliferation and survival, including the RAS/ERK pathway. The activation of these pathways due to AURKA overexpression can lead to increased cell cycle progression and resistance to apoptosis, ultimately contributing to treatment failure. 63
Given the pivotal role of AURKA in resistance mechanisms, AURKA inhibitors have surfaced as a promising therapeutic strategy to counteract this challenge. Preclinical studies have demonstrated that inhibiting AURKA can restore sensitivity to CDK4/6i in cell lines with AURKA amplification, indicating the potential for combination therapies. 64 The selective AURKA inhibitor LY3295668 demonstrated efficacy in a phase I monotherapy safety study, achieving a disease control rate of 69% of the 12 patients with locally advanced solid tumors. 65 A phase II trial assessed alisertib with or without fulvestrant in 91 postmenopausal women with endocrine-resistant and CDK4/6i-resistant MBC. Response rates were similar for both treatment arms (approximately 20%), with alisertib monotherapy demonstrating activity; however, the combination did not improve outcomes. 66
Hyperactivation of the PI3K/AKT/mTOR signaling pathway
The PI3K/AKT/mTOR signaling pathway represents a vital cellular network that regulates growth, metabolism, and cell survival (Figure 1). 67 In HR+ breast cancer, the hyperactivation of this pathway has emerged as a significant contributor to resistance against standard treatment, as CDK4/6i in particular. 68 Approximately 50% of HR+/HER2− MBC cases show alterations in this pathway, including mutations in the PI3KCA gene and loss of function of the tumor suppressor PTEN. Such alterations enhance cyclin D expression and activity, which ultimately reduces the efficacy of CDK4/6i. 69
Clinical investigations consistently demonstrate that alterations in the PI3K/AKT/mTOR pathway are associated with poorer survival outcomes for patients with HR+ MBC, with PIK3CA mutations or PTEN loss correlating with decreased PFS and OS following treatment with ET and CDK4/6i. 70 Studies indicate that patients with ET-resistant disease exhibit increased activation of the PI3K/AKT/mTOR pathway before initiating CDK4/6i treatment, highlighting the pathway’s role in resistance development. 71 However, it is crucial to differentiate between prognostic and predictive biomarkers in this context. While alterations in the PI3K/AKT/mTOR pathway serve as prognostic biomarkers that provide insights into disease progression and overall patient outcomes, they do not function as predictive biomarkers for guiding treatment decisions regarding CDK4/6i therapy. Thus, although these alterations indicate a worse prognosis, they do not inform oncologists about the likely efficacy of CDK4/6i for individual patients.
The introduction of specific inhibitors targeting the PI3K/AKT/mTOR pathway has significantly reshaped the treatment landscape for patients with HR+ advanced breast cancer, mostly for those who have progressed following prior ET. 72 Among the new FDA-approved agents, alpelisib (a selective PI3Kα inhibitor) and capivasertib (an AKT inhibitor) have demonstrated considerable efficacy against tumors harboring PIK3CA mutations or showing elevated AKT signaling. Clinical trials indicate that the combination of alpelisib with ET substantially improves PFS in patients with PIK3CA-altered breast cancer, resulting in clinically meaningful responses even within heavily pre-treated populations.73,74 Conversely, capivasertib has shown potential in countering multiple resistance mechanisms, as those driven by AKT hyperactivation, mainly when used alongside endocrine agents. 75
Everolimus, a well-known mTOR inhibitor, significantly enhanced median PFS by 64% within the BOLERO-2 trial when combined with exemestane in later line of therapy. 76 In a phase III trial, inavolisib, a selective PI3K inhibitor, combined with palbociclib and fulvestrant, improved PFS in patients with HR+/HER2− PIK3CA-mutated advanced breast cancer, with a median of 15.0 versus 7.3 months in the placebo group (hazard ratio = 0.43, 95% confidence interval: 0.32–0.59, p < 0.001). The inavolisib group had a higher incidence of grade 3 or 4 adverse events, but treatment discontinuation due to these complications was low. 77
Recent developments in the treatment of HR+ breast cancer have brought several promising PI3K/AKT/mTOR inhibitors to the forefront. Buparlisib, the first pan-PI3K inhibitor to enter global phase III trials, showed a modest improvement in PFS in the BELE-2 and BELLE-3 studies, but also exhibited notable toxicity, including elevated liver enzymes and hyperglycemia.78,79 Pictilisib is another orally bioavailable pan-PI3K inhibitor that was assessed in the phase II FERGI trial; however, it failed to show significant differences in PFS against placebo, accompanied by a high incidence of grade 3 or 4 adverse events. 80 Taselisib, a selective inhibitor targeting class I PIK3CA isoforms, demonstrated modest improvements in investigator-assessed PFS in patients with PIK3CA mutations during the phase III SANDPIPER trial but also faced considerable treatment-related adverse events like diarrhea and hyperglycemia. 81
In the setting of AKT inhibitors, Ipatasertib has been evaluated in the IPATunity130 trial but did not improve PFS or objective response rates among patients with PI3K pathway mutations. 82 Gedatolisib, a dual inhibitor of class I PI3Ks and mTOR, exhibited a compelling overall response rate in a phase Ib trial when used with palbociclib and ET. 83 Ongoing studies are exploring the efficacy and safety of both gedatolisib and inavolisib in combination therapies for advanced HR+ breast cancer. A myriad of novel agents are being studied in early-phase clinical trials with promising results concerning improved tolerability and targeted action in the future.84–87
These data show that, despite the unquestionable efficacy of combined PI3K/AKT/mTOR–CDK4/6i therapies in HR+ MBC, PI3K/AKT/mTOR inhibitors are not interchangeable in terms of efficacy and tolerability—with many of them proving unfeasible for either single agent or combination strategies. Therefore, safety, quality of life, and patient-reported outcomes (instead of efficacy alone) should be key elements on the interpretation of studies with these agents.
This toxicity profile that affects the clinical use of PI3K/AKT/mTOR inhibitors in HR+ MBC exhibits significant variability. Alpelisib, while effective, has a high incidence of adverse events, with distinction to hyperglycemia, which impacted around 63.7% of patients and was classified as grades 3 or 4 in 36.6% cases during pivotal trials like SOLAR-1. 88 In contrast, capivasertib shows a more favorable safety profile, with only 16.3% of patients experiencing hyperglycemia and 2.3% reporting grade 3 or 4 hyperglycemia in the CAPItello-291 trial. 89 Emerging isoform-selective and mutant-selective inhibitors in phase I trials offer the potential for effective antitumor activity with fewer metabolic side effects. To enhance tolerability, strategies include using these selective agents to lessen on-target toxicity, employing alternative dosing regimens, and integrating hypoglycemic agents like metformin to manage hyperglycemia. 72
RAS/MAPK hyperactivation
The activation of the MAPK pathway, mainly through components such as NF1, KRAS, BRAF, and MAP2K1, plays a significant role in endocrine resistance in breast cancer. 90 Mutations in these genes are rare in primary early-stage breast cancers, and more frequent in MBC, especially in NF1, which negatively regulates RAS activity. 91 The loss of NF1 function leads to hyperactivation of RAS and contributes to both intrinsic and acquired resistance to ET. Preclinical studies have shown that NF1 loss promotes ligand-independent expression of cyclin D1 and enhances resistance to CDK4/6i (Figure 1), further impairing treatment strategies in HR+ breast cancer. 92
The ERK pathway is intricately involved in ER signaling, with non-genomic actions of estrogen resulting in the rapid activation of RAS and downstream pathways. This activation enhances the proliferative and survival effects of estrogen, while also leading to the overexpression of receptor tyrosine kinases (RTKs) that further promote endocrine resistance. Activation of the PI3K and ERK pathways by RTKs and ER itself results in the phosphorylation of ER coregulators, enhancing ER activity and contributing to resistance mechanisms. 93 Evidence indicates that phosphorylation of RAS, RAF, and ERK correlates with poor outcomes in patients treated with ET like tamoxifen, highlighting the clinical impact of MAPK pathway activation. 94
Despite promising preclinical evidence of using ERK pathway inhibitors to overcome endocrine resistance, clinical outcomes have not consistently supported these approaches. 95 A trial evaluating the combination of fulvestrant with selumetinib, a MEK inhibitor, found no significant benefits in enhancing treatment response. 80 ctDNA analyses have identified RAS mutations in a subset of patients with disease progression under aromatase inhibitors (AI), underscoring the complexity of resistance mechanisms linked to MAPK pathway activation. 96
“Non-LUMINAL” intrinsic subtypes
In preclinical and clinical scenarios, luminal breast cancers are sensitive to CDK4/6i. This observation is supported by preclinical data sets from HR+ breast cancers, TNBC, and HER2+ disease. 82 However, the efficacy of CDK4/6i in non-luminal HR+/HER2− breast cancer remains unclear. Immunohistochemistry-based classifications and intrinsic subtypes show a moderate correlation but are not identical. 97
A notable proportion of HR+ tumors (5%–20%) may exhibit non-luminal characteristics, which could suggest a potential reduction in sensitivity to ET and an increased likelihood of responsiveness to chemotherapy, thereby possibly affecting the overall prognosis. Furthermore, tumor progression has been associated with a transition toward a more aggressive, ER-independent, non-luminal phenotype. 98 A retrospective analysis of biomarkers related to intrinsic subtypes and the response to treatment within the MONALEESA phase III clinical trials, which included 1160 patients, showed that most breast cancer subtypes significantly benefited from adding CDK4/6i to ET. However, the basal-like subtype did not exhibit this benefit, with a median PFS of 3.7 months for those receiving ribociclib compared to 3.5 months for those on placebo. 99
Notably, the HR+ basal tumors (n = 309, 2.6%) in this cohort exhibited high expression levels of cyclin E1 and epidermal growth factor receptor, along with low expression of luminal-related genes, resembling the TNBC phenotype. Another interesting finding was that the HR+/HER2− with HER2-enriched subtype had a poorer prognosis in the ET alone arms but derived a more significant benefit from the addition of CDK4/6i, with a considerable proportion of initially HER2-enriched tumors converting to the luminal A subtype upon rebiopsy. This translational study offers a rationale for employing intrinsic subtyping of breast cancers for the prospective allocation of CDK4/6i combination therapies in future clinical research contexts. 99
The benefit of CDK4/6i in patients with non-luminal intrinsic subtypes warrants further investigation, and an international phase II clinical trial has been designed to evaluate the safety and efficacy of trastuzumab deruxtecan (T-DXd) compared to CDK4/6i-based ET as 1L therapy for HR+ and HER2-low/ultralow MBC patients classified as non-luminal subtype by PAM50 (PONTIAC; NCT06486883). Interestingly, a meta-analysis found that HER2-low status was not significantly associated with Objective Response Rate (ORR) or OS in patients receiving later line or 1L CDK4/6i with ET. 100 A secondary analysis of the PALOMA-2 and PALOMA-3 trials indicated that CDK4/6i combined with ET improved PFS in HER2-low-positive patients, while benefits in HER2-0 patients were mainly observed in those who had progressed on prior ET. 101 Another prospective study also failed to validate HER2-low status as a predictive biomarker for the use of CDK4/6i plus ET in HR+ MBC. 102
Pathogenic germline mutations
The co-occurrence of germline and somatic oncogenic alterations is frequently observed in breast cancer; however, their combined biological and clinical significance has not been thoroughly evaluated. Recent studies suggest that pathogenic germline mutations, with attention to germline BRCA1/2 (gBRCA1/2) mutations, may play a dual role in influencing the efficacy of CDK4/6i. 103 In one hand, BRCA2-mutant tumors often exhibit increased genomic instability, which may sensitize them to combination strategies involving CDK4/6i and DNA-damaging agents. On the other hand, these mutations can also lead to adaptive resistance mechanisms by rewiring cell cycle machinery, potentially reducing the efficacy of CDK4/6i as monotherapy.104,105
To assess the impact of germline–somatic interactions on outcomes in routine practice, Safonov et al. 106 developed an integrated clinic-genomic pipeline to analyze the genomes of over 4500 breast cancer patients. They found that gBRCA2-associated tumors are enriched with RB1 loss-of-function mutations and show poor outcomes with 1L CDK4/6i. Among these tumors, gBRCA2-related homologous recombination deficiency (HRD) and baseline RB1 LOH status facilitate the acquisition of RB1 loss-of-function mutations under the selective pressure of CDK4/6i, leading to therapy resistance. These findings suggest an alternative therapeutic strategy that involves sequentially targeting HRD in gBRCA-associated breast cancers using PARP inhibitors prior to CDK4/6i therapy to mitigate deleterious RB1 loss trajectories and suppress the development of CDK4/6i resistance. More broadly, these significant findings illustrate how germline-somatic driven genomic configurations influence responses to systemic therapy and can be leveraged therapeutically by biomarker-directed clinical practice strategies.
An increasing body of real-world evidence suggests that patients with germline pathogenic variants in DNA damage repair-related genes, as BRCA1, BRCA2, and PALB2, may experience suboptimal outcomes with CDK4/6i.107–112 These findings were further supported by a recent meta-analysis, which demonstrated that germline BRCA carriers with HR+/HER2− MBC treated with CDK4/6i combined with ET had significantly worse PFS and OS, with a two-fold risk of death compared to BRCA non-carriers. 113 In an exploratory analysis of the MonarchE trial, 3.5% of patients had gBRCA1/2 mutations, with only 1 of 20 (5%) on abemaciclib plus ET experiencing an invasive disease-free survival (iDFS) event, compared to 9 of 21 (42.8%) on ET alone. 114
To address this unmet need, the phase III randomized EvoPAR-Breast01 trial (NCT06380751) will evaluate the efficacy of saruparib (a next-generation PARP1-selective inhibitor) combined with camizestrant (a next-generation oral selective estrogen receptor degrader (SERD)) compared to the clinician’s choice of CDK4/6i plus ET (or oral SERD) as 1L treatment for patients with advanced-stage ER-positive (ER+)/HER2− breast cancer harboring BRCA1, BRCA2, or PALB2 mutations.
Table 1 summarizes key clinical trials in HR+ breast cancer that reported findings on biomarkers associated with resistance to CDK4/6i.
Key clinical trials published that investigate potential biomarkers of resistance to CDK4/6i.

ABC, advanced breast cancer; AKT, protein kinase B; AURKA, aurora kinase; CBR, clinical benefit rate; CCNE1, cyclin E1 gene; CDK4/6i, cyclin-dependent kinase 4/6 inhibitor; ctDNA, circulating tumor DNA; ER+, estrogen receptor-positive; ET, endocrine therapy; FGFR, fibroblast growth factor receptor; HER2−, human epidermal growth factor receptor 2-negative; MBC, metastatic breast cancer; mTOR, mammalian target of rapamycin; ORR, objective Response Rate; PFS, progression-free survival; PI3K, phosphatidylinositol 3-kinase.
Future directions and emerging therapeutic opportunities in clinical practice
The future of treatment for HR+/HER2− MBC is poised to transform, driven by continuous advancements in clinical practices and genomic research. The focus of this evolution encompasses some critical areas:
A more profound understanding of clinical and genomic characteristics that can help identify patients eligible for alternative treatment strategies beyond 1L ET.
Validation of the clinical utility of ctDNA monitoring, facilitating early interventions for disease progression and enhancing patient outcomes.
An in-depth exploration of resistance mechanisms that will pave the way for the creation of highly effective combination therapeutic strategies.
Table 2 presents an overview of ongoing clinical trials in HR+ MBC that explore proposed strategies aimed at overcoming resistance to CDK4/6i.
Ongoing clinical trials exploring strategies to overcome resistance to CDK4/6i.

1L, first-line; ADC, antibody-drug conjugate; AKT, protein kinase B; CDK4/6i, cyclin-dependent kinase 4/6 inhibitors; ctDNA, circulating tumor DNA; ET, endocrine therapy; HER2, human epidermal growth factor receptor 2; HR, hormone receptor; MBC, metastatic breast cancer; mTOR, mammalian target of rapamycin; ORR, objective Response Rate; PFS, progression-free survival; SERD, selective estrogen receptor degrader; T-DXd, trastuzumab deruxtecan.
Innovations addressing primary resistance to CDK4/6i
Approximately 10% of patients exhibit primary resistance to CDK4/6i, reaping minimal benefits from this therapeutic strategy. An exploratory single-cell RNA sequencing study suggested that CD8+ T and NK cells may serve as baseline response predictors, with higher levels of tumor-infiltrating CD8+ T and NK cells found in long-term responders. 123 Razavi et al. developed a machine learning (ML) model that utilizes both clinical and genomic baseline characteristics at the time of metastatic recurrence to predict the outcomes of 1L CDK4/6i combined with ET. This multimodal ML tool exhibited superior prognostic and predictive capabilities compared to traditional models reliant solely on clinical or genomic data. 124
The ML model stratified patients into four risk groups, showing markedly different median PFS: 5.3 months in the high-risk group, 29 months in the low-risk group, and 10.7 and 19.8 months in the intermediate-risk groups. These findings highlighted the significant heterogeneity in response to 1L CDK4/6i and the clinical need to identify patients unlikely to benefit from this therapy. Key clinical and genomic features used by the multimodal model included tumor mutational burden, fraction of genome altered (FGA), TP53 alterations, the fraction of the genome with LOH, presence of liver metastasis, adjuvant treatment-free interval of <1 year, primary tumor grade 3, presence of visceral metastasis, primary tumor progesterone receptor negativity, and whole genome doubling. Importantly, specific actionable alterations in several genes, such as PTEN, AKT, ESR1, ERBB2, somatic BRCA1/2, and TP53 mutations, were more prevalent in high-risk groups. These findings highlight potential avenues for emerging strategies in this challenging clinical setting. 125
The GEICAM/2014-03 RegistEM registry trial showed a median PFS of 8.1 months for 1L treatment with CDK4/6i plus ET offered to a subgroup of patients with HR+ MBC exhibiting primary resistance to ET—defined as relapse within the first 2 years of adjuvant ET. The presence of PIK3CA mutations was associated with a shorter response in this specific cohort. In another subgroup classified as endocrine-sensitive, based on recurrence after 12 months of completion of the prior adjuvant ET, patients who progressed within the first 6 months of 1L treatment with CDK4/6i plus ET experienced poorer OS. 126 By contrast, a sub analysis of the Destiny Breast-06 trial regarding the pace of disease progression on prior ET-based therapy indicated that the median PFS for patients who progressed within the first 6 months on 1L ET plus CDK4/6i was 14 months. 127 These collective data contribute to the identification and characterization of patient subgroups that exhibit intrinsic resistance to CDK4/6i in the 1L treatment setting, requiring the development of alternative therapeutic strategies.
Emerging approaches in the scenario of clinical utility of ctDNA monitoring
In metastatic setting, the clinical utility of ctDNA for the detection of targetable genomic alterations has been demonstrated. Tumor next-generation sequencing by tissue or plasma sample is recommended for endocrine-resistant HR+ MBC. 128 The serial collection of ctDNA has the potential to predict treatment efficacy either by monitoring ctDNA kinetic and dynamic assessing early drug response and resistance or by detecting the early rise of established resistance variants that predict poor outcomes. The short half-life of ctDNA offers a unique opportunity to utilize its timely on-treatment changes for real-time assessment of therapeutic response and outcomes, named molecular response. 129
The molecular response would allow early identification of patients who will derive clinical benefit to continue therapy while avoiding unnecessary health and financial toxicities by enabling re-stratification to more effective therapeutic strategies of those with early prediction of resistance and treatment failure. However, critical questions remain unanswered, such as the optimal method and time points for calculating ctDNA molecular response, and future prospective trials are warranted. Accumulating data in MBC have suggested the great potential of baseline and early dynamics in serial ctDNA monitoring for prognostic assessment and for prediction of treatment response or resistance. 130
Retrospective analysis and prospective data have demonstrated that early ctDNA changes in the first 2 weeks on 1L CDK4/6i-based therapy were strongly associated with different patterns of clinical response as well as PFS and OS.131,132 Monitoring ctDNA dynamics to determine molecular progression of disease and the utility of switching therapy before clinical progression is an ongoing investigational strategy in different clinical trials (Table 2). SAFIR 03–ARRIBA (NCT05625087) is evaluating the clinical utility of early therapy switch for endocrine-resistant, PIK3CA mutated, HR+/HER2− MBC on 1L CDK4/6i plus Fulvestrant strategy with PIK3CA ctDNA level persistent after 4 weeks of treatment. Patients with a high risk of relapse will be randomized to Alpelisib plus Fulvestrant or Ribociclib plus Fulvestrant. Another study from the same group, SAFIR 03–LibHERty (NCT06680596), will evaluate the benefit of an early switch to T-DXd (single arm) for endocrine-resistant and sensitive, HR+/HER2-low or ultra-low MBC on 1L CDK4/6i-based therapy with ctDNA persistent at 4 weeks of treatment (ClinicalTrials.gov identifier: NCT03386162).
The PADA-1 study evaluated the efficacy of an early therapy switch based on a rising ESR1 that encodes the ER ligand-binding domain (referred to as ESR1 mutations (ESR1m)), which causes constitutive (estrogen-independent) activation of ER. This mutation detection was performed by ctDNA without radiologic disease progression in patients treated with 1L palbociclib plus AI. Those patients were randomized to continue AI versus switching to fulvestrant while continuing palbociclib. The median PFS from random assignment was 11.9 and 5.7 months in favor of fulvestrant plus palbociclib. PADA-1 is the first trial to show the clinical utility and feasibility of real-time serial ctDNA monitoring and the efficacy and safety of a circulating biomarker-driven early switch versus an imaging-driven switch at tumor progression. In the optional crossover cohort of the trial, the strategy of using fulvestrant after clinical tumor progression on AI plus CDK4/6i showed a short PFS2 of 3.5 months. 133
The results of PADA-1 suggest the clinical benefit of targeting resistance mutations earlier when the burden of resistant tumor cells is still low and with fewer polyclonal resistance mechanisms. 134 However, ESR1m are heterogeneous in terms of response to treatment, and some variants, such as the Y537S ESR1, might be less sensitive to fulvestrant, and the next-generation oral SERDs could further expand the clinical benefit observed in this trial. 135 Applying the same principle, the ongoing SERENA-6 study (NCT049649) is currently exploring the benefit of a switch from an AI to the next-generation oral SERD camizestrant as the endocrine backbone to CDK4/6i upon the first detection of ESR1m in ctDNA. 136
Emerging combined therapeutic strategies in the first-line setting based on the comprehensive characterization of resistance mechanisms
The prolonged exposure to ET will eventually lead to the development of resistance mechanisms, which can be classified as ER-dependent or ER-independent. Therefore, this evidence of divergent tumor evolutionary routes in single patients highlights the relevance of combination approaches to HR+/HER2− MBC treatment. Preclinical research showed that substantial synergy can be achieved with simultaneous blockade of the key oncogenic drivers of ER+ breast cancer in xenograft models by further reducing the tumor burden and preventing or delaying the emergence of resistance to treatment. 137
The previously mentioned phase III INAVO 120 trial has demonstrated that the triplet therapy regimen—with the addition of inavolisib to standard doublet regimen of palbociclib/fulvestrant—resulted in statistically and clinically significant longer PFS and OS in patients with PIK3CA-mutated, HR+/HER2− MBC whose disease had recurred during or within 12 months after the completion of adjuvant ET, including one-third of patients with primary endocrine resistance with an acceptable level of toxicity. 77 Recently, the IPATunity study (NCT04060862) that will evaluate the efficacy of Ipatasertib/Palbociclib/Fulvestrant compared to Palbociclib/Fulvestrant in the same clinical setting was interrupted before initiation of phase III as per sponsor’s decision. The Morpheus-panBC (NCT03424005) and Capitello-292 (NCT04862663) studies will evaluate the triplet combinations with Inavolisib and Capivasertib, respectively, in the 1L setting. Many early-phase clinical trials in the later lines are evaluating doublet or triplet combinations involving tyrosine kinase, mutant-selective PIK3, Pan-FGFR kinase, CDK 2 and 7, and AURKA inhibitors. Figure 2 highlights potential windows of opportunity for designing clinical studies that could lead to transformative changes in practice and Figure 3 identifies opportunities for clinical practice changes that could lead to significant timely better patient care.

Windows of opportunity for new practice-changing clinical studies designs.
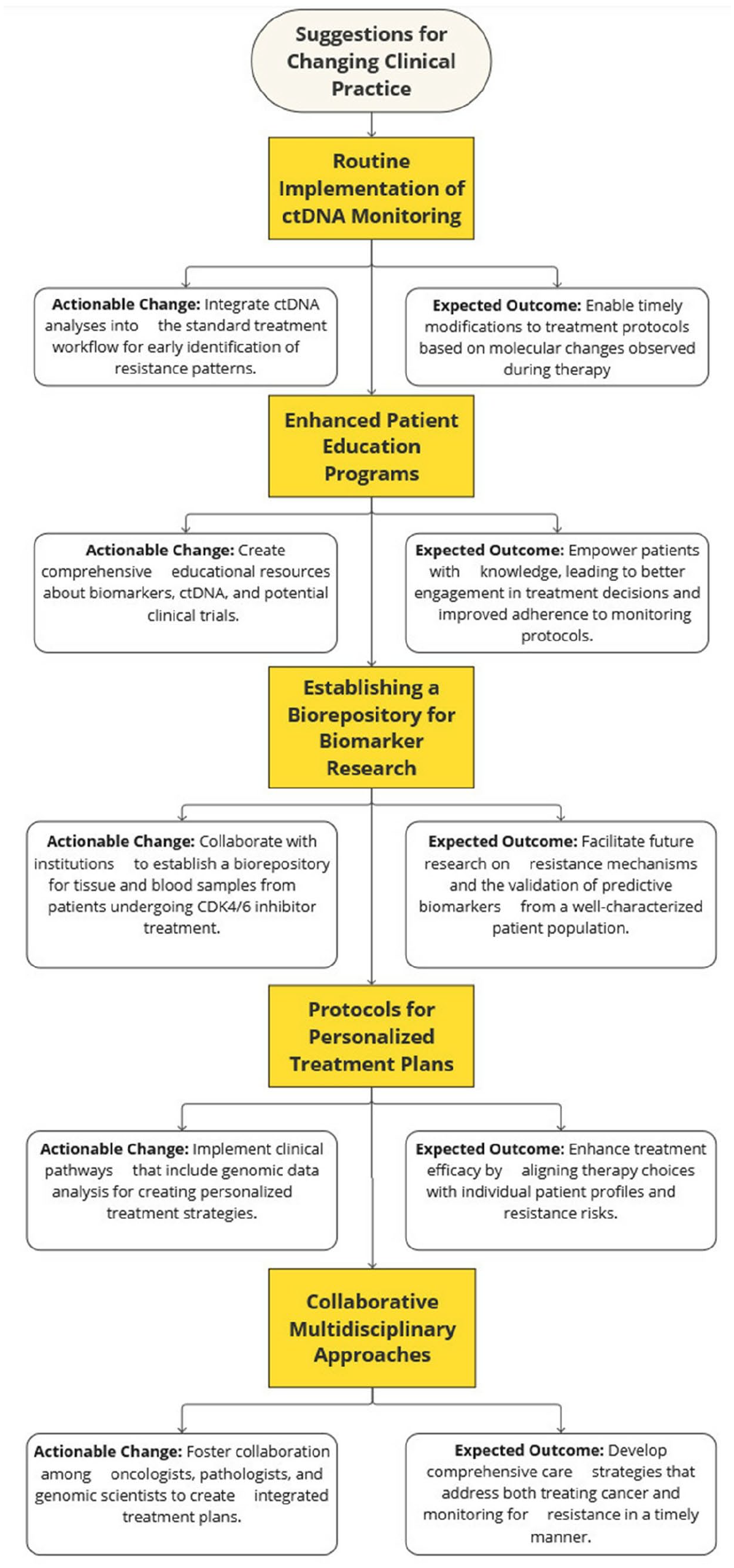
Suggestions of timely accessible clinical practice changes that can foster better patient care and a promisor environment for scientific development.
Conclusion
Despite significant advances in treating HR+ MBC, resistance to CDK4/6i remains a formidable challenge that impairs durable responses and hinders prognosis. The complexity of resistance mechanisms, such as mutations in the RB1 and PIK3CA genes, as well as the dysregulation of CDK2 and cyclin E, underscores the importance of developing targeted biomarker strategies to predict treatment outcomes. Furthermore, patients may experience intrinsic resistance to the 1L combination of CDK4/6i with ET, resulting in shortened survival, while exposure to this regimen can lead to acquired resistance. Future research must prioritize key areas that comprise: (1) thoroughly elucidating the mechanisms of action of CDK4/6i; (2) understanding their synergy with the various available ET; (3) optimizing rescue treatment options following disease progression on CDK4/6i plus ET; (4) investigating the resistance mechanisms to CDK4/6i plus ET. Finally, the integration of ctDNA for real-time monitoring could enable early detection of acquired resistance and facilitate personalized treatment strategies. By prioritizing these critical areas, we can improve treatment efficacy and patient outcomes in the challenging landscape of HR+ MBC.