
Abstract
Deregulation of the cell cycle is a hallmark of cancer, and research on cell cycle control has allowed identification of potential targets for anticancer treatment. Palbociclib is a selective inhibitor of the cyclin-dependent kinases 4 and 6 (CDK4/6), which are involved, with their coregulatory partners cyclin D, in the G1-S transition. Inhibition of this step halts cell cycle progression in cells in which the involved pathway, including the retinoblastoma protein (Rb) and the E2F family of transcription factors, is functioning, although having been deregulated. Among breast cancers, those with functioning cyclin D-CDK4/6-Rb-E2F are mainly hormone-receptor (HR) positive, with some HER2-positive and rare triple-negative cases. Deregulation results from genetic or otherwise occurring hyperactivation of molecules subtending cell cycle progression, or inactivation of cell cycle inhibitors. Based on results of randomized clinical trials, palbociclib was granted accelerated approval by the US Food and Drug Administration (FDA) for use in combination with letrozole as initial endocrine-based therapy for metastatic disease in postmenopausal women with HR-positive, HER2-negative breast cancer, and was approved for use in combination with fulvestrant in women with HR-positive, HER2-negative advanced breast cancer with disease progression following endocrine therapy. This review provides an update of the available knowledge on the cell cycle and its regulation, on the alterations in cyclin D-CDK4/6-Rb-E2F axis in breast cancer and their roles in endocrine resistance, on the preclinical activity of CDK4/6 inhibitors in breast cancer, both as monotherapy and as partners of combinatorial synergic treatments, and on the clinical development of palbociclib in breast cancer.
Keywords
Introduction
Abnormal cell proliferation, often resulting from oncogene activation or from inactivation or deletion of tumor-suppressor genes, is the key hallmark of cancer [Hanahan and Weinberg, 2011], and the main players of cell cycle control are logical potential targets for cancer therapy.
Breast cancer is the most common malignancy in women worldwide, and the most common cause of cancer death in women in less developed regions, and the second in women in more developed regions [International Agency for Research on Cancer, 2016]. Although breast cancer death rates are decreasing in developed countries, metastatic breast cancer remains largely an incurable disease, and new treatments are needed.
Palbociclib is the first of a new class of agents targeting the cell cycle machinery.
We will review recent knowledge on the cell cycle and its regulation, on the alterations in key cell cycle molecules in breast cancer and their roles in endocrine resistance, on the preclinical activity of cyclin-dependent kinases 4 and 6 (CDK4/6) inhibitors and on clinical development of palbociclib in breast cancer. The relevant literature was retrieved through PubMed (main keywords used: CDK 4/6 inhibitor, palbociclib, PD 0332991, ribociclib, abemaciclib; time period: up to May 2016) and the proceedings of the main recent cancer congresses (ASCO and AACR annual meetings 2014–2016, San Antonio Breast Cancer Symposium 2014–2015).
Cyclin-dependent kinases and the cell cycle
Major players of the cell cycle are the serin/threonin kinases CDKs [Malumbres, 2014], their activating regulatory subunits cyclins [Ma et al. 2013], the so-called pocket proteins retino-blastoma (Rb, product of the Rb tumor-suppressor gene RB1) and Rb-like proteins p107 (RBL1) and p130 (RBL2) [Dick and Rubin, 2013], and the E2F family of transcription factors [Chen et al. 2009] (Figure 1). The different cyclins, each synthesized cyclically in specific phases of the cell cycle, bind to, and activate, specific CDKs, and the cyclin-CDK complexes govern the progression of the cell throughout the cycle, a process whose main limiting step is inhibiting Rb and thus releasing its inhibitory action on E2F, allowing transcription of genes that execute the cell cycle.

The cyclin D–cyclin-dependent kinase 4/6–retinoblastoma protein–E2F transcription-factor axis.
Two main classes of CDK inhibitors (cyclin kinase inhibitors, CKI) exist: the inhibitor of CDK4 (INK4) family, including p16INK4A, p15INK4B, p18INK4C, and p19INK4D, specifically blocking the formation of cyclin D-CDK4/6 complexes [Sherr and Roberts, 1999], and the CDK-interacting protein/kinase inhibitory protein (CIP/KIP) family, including p21Cip1, p27Kip1, and p57Kip2, acting on all cyclin-CDK complexes, with prevalent inhibitory, but sometimes activatory effects, in particular, activating cyclin D-CDK4/6 complexes [LaBaer et al. 1997]. CDK activators, such as the cell-division cycle 25 (Cdc25) phosphatase family, also exist [Shen and Huang, 2012].
In the established model of cell cycle regulation in mammalians [Malumbres and Barbacid, 2009], mitogenic stimuli induce the expression of D-type cyclins (D1, D2, and D3), that bind to the two closely related CDKs 4 and 6, collectively called CDK4/6 for their similar function, activating their kinase activity. Cyclin D–CDK4/6 complexes phosphorylate Rb (and the other pocket proteins) on specific serine and threonine residues. Rb is active when unphosphorylated, its main function being binding to and blocking the activity of transcription factors, mainly of the E2F family, responsible for the transcription of genes involved in cell cycle progression [Weinberg, 1995]. Phosphorylation of Rb leads to its inactivation, with consequent release of E2F transcription factors that start their activity, promoting progression of the cell through the division cycle. Progressive phosphorylation of Rb occurs during the G1 (gap 1) phase of the cycle, culminating at the ‘restriction point’ (R point), when an ‘all or none’ decision is made on cell fate, depending on the balance between growth-promoting and growth-inhibitory stimuli, leading to G1-S transition and progression through the subsequent phases of the cycle, or to the quiescent state, G0.
Progression through the R point is pushed by a positive feedback, elicited by E2F transcription of cyclin E and the CDK activators Cdc25 phosphatases, leading to formation of cyclin E–CDK2 complexes, which further phosphorylate the pocket proteins [Lundberg and Weinberg, 1998; Harbour et al. 1999]. Another forward mechanism is due to the CIP/KIP proteins, which, during G1, bind to and activate the increasing cyclin D–CDK4/6 complexes, being therefore subtracted from cyclin E–CDK2 complexes (on which they are inhibitory), favoring their activation [LaBaer et al. 1997]. Moreover, cyclin E–CDK2 complexes phosphorylate p27Kip1, leading to its degradation [Slingerland and Pagano, 2000]. The conjunct action of cyclin D–CDK4/6 and cyclin E–CDK2 complexes leads to progression of the cell cycle beyond the R point, from the G1 to the S phase.
CKI, which are silenced during the cell cycle, are expressed in response to inhibitory signals such as transforming growth factor β [Reynisdóttir et al. 1995], contact inhibition [Polyak et al. 1994], or senescence [Sharpless and Sherr, 2015].
Emerging concepts in the cell cycle and key molecules’ roles
Recent evidences have led to a widened picture of cell cycle control and of its key molecules’ roles.
Although being major players in cell cycle, CDK4/6 and CDK2 are generally dispensable for cycling of most mammalian cells, being required only in specific cell types [Malumbres et al. 2004; Malumbres and Barbacid, 2009]. D-type cyclins can bind different CDKs to initiate the cell cycle [Xiong et al. 1992]. Only CDK1, which binds cyclin A during the G2 phase and cyclin B for entry into mitosis, is indispensable and sufficient for driving the cell cycle in all cell types during embryogenesis [Santamaría et al. 2007]. Nonetheless, some tumor cells may acquire dependency on other CDKs for proliferation [Malumbres and Barbacid, 2009]. In mouse models, cyclin D1 [Yu et al. 2001] and CDK4 [Reddy et al. 2005; Landis et al. 2006; Yu et al. 2006] are necessary for the development of breast cancers induced by the neu and ras oncogenes, but not for those driven by c-Myc or Wnt-1, highlighting the dependency of certain tumors from specific cyclin–CDKs, which can be exploited therapeutically [Choi et al. 2012]. The dependency on cyclin D1 can nonetheless be overcome by cyclin D3 overexpression, in case of cyclin D1 loss [Zhang et al. 2011].
The levels of CDK2 at the exit from mitosis, which are regulated by mitogens via p21 at the end of the cell cycle, are important in deciding cell fate, leading to further proliferation when CDK2 is elevated, or to a quiescent state when suppressed [Spencer et al. 2013].
Also the G0 to G1 transition requires Rb phosphorylation, resulting from cyclin C binding to and activating CDK3 [Ren and Rollins, 2004].
Cyclins and CDKs have several functions beyond their canonical role in cell cycle [Hydbring et al. 2016; Musgrove et al. 2011]. Cyclin D–CDK complexes phosphorylate several transcription factors, and regulate their function independently of their action on the pocket proteins. CDK4 and CDK2 phosphorylate and inhibit small mother against decapentaplegic 3 (SMAD3), a key mediator of the anti-proliferative activity of transforming growth factor-β, favoring cell cycle progression from G1 to S phase [Matsuura et al. 2004]. Cyclin D–CDK4/6 complexes phosphorylate and activate the forkhead box protein M1 that sustains the expression of cell cycle genes and protects cancer cells from senescence [Anders et al. 2011]. Cyclin D1/CDK4 complexes phosphorylate methylosome protein 50 leading to protein arginine methyltransferase 5-dependent histone methylation and transcriptional repression of CUL4, which results in overexpression of the replication-licensing protein CDT-1, required for initiation of DNA replication [Aggarwal et al. 2010]. Cyclin D–CDK6 complexes, through activation of JUN and signal transducer and activator of transcription 3, induce the expression of p16INK4A, exerting a negative feedback control of their own activity [Kollmann et al. 2013]. Indeed, the transforming effect of CDK6 overexpression may occur only upon deletion or silencing of CDKN2A, the gene encoding p16INK4A that frequently occurs in breast cancer [Cancer Genome Atlas Network, 2012]. CDK6 cooperates with the NF-κB subunit p65 to induce the expression of inflammatory genes [Handschick et al. 2014]. Cyclin D–CDK4/6 complexes are also involved in glucose metabolism [Lee et al. 2014] and in the processes of apoptosis and cell differentiation [Hydbring et al. 2016]. The relevance of all these effects in different tumor types and their implication for the activity of CDK4/6 inhibitors must be better clarified.
Cyclins and CDKs can also regulate transcription factors and exert other functions independently from cyclin–CDK complexes’ kinase activity [Hydbring et al. 2016], and therefore not subject to inhibition by current CDK4/6 inhibitors. Among these are: a direct role of cyclin D1 in gene transcription [Bienvenu et al. 2010]; its involvement in DNA damage repair [Jirawatnotai et al. 2011]; its repression of specificity protein 1-mediated transcription [Shao and Robbins, 1995]; inhibition of cyclin D-interacting myb-like protein (DMP1, affecting, via p19ARF and MDM2, the expression of p53) [Hirai and Sherr, 1996]; induction of vascular endothelial growth factor (VEGF) expression [Yasui et al. 2006]; its activating action on the estrogen receptor (ER) [Zwijsen et al. 1997], and inhibitory action on the androgen receptor [Reutens et al. 2001]. CDK4/6 also regulates the expression of VEGF [Abedin et al. 2010; Kollmann et al. 2013] and is involved in DNA repair [Dean et al. 2012a].
Beyond Rb, the Rb-like proteins p130 and p107 have a role in repressing cell cycle gene expression during quiescence and in coordinating gene expression during S phase and G2/M phase [Sadasivam and DeCaprio, 2013]. However, mutations are much more frequent in Rb gene than in genes of the other family proteins, and Rb has a nonredundant role in repressing E2F target genes involved in DNA replication during cell senescence [Chicas et al. 2010].
Accumulating evidence also shows roles for the E2F transcription factors beyond that in the cell cycle [Johnson et al. 2016], including roles in promoting invasiveness [Yoon et al. 2006] and metastatization [Andrechek, 2015].
Cyclin–cyclin-dependent kinase–retinoblastoma protein–E2F pathway alterations in breast cancer
Genomic aberrations or altered expression of molecules of the cyclin–CDK–Rb pathway are frequent in breast cancer. According to The Cancer Genome Atlas [TCGA; Cancer Genome Atlas Network, 2012], the gene encoding for cyclin D1 (CCND1) is amplified in 29% and 58% of luminal A and B tumors, respectively, and in 38% of HER2-enriched tumors, whereas that encoding cyclin E (CCNE1) is amplified in 9% of triple-negative tumors. Cyclin D1 overexpression, detected by immunohistochemistry in up to half of breast cancers, is even more frequent than CCND1 amplification, implying that other mechanisms can lead to deregulated expression [Bartkova et al. 1994; Gillett et al. 1994].
Amplification of the CDK4 gene is reported in about 15% of breast cancers, results in protein overexpression, and is associated with high Ki-67 labeling index [An et al. 1999]. It occurs in 14% of luminal A, 25% of luminal B and 24% of HER2-enriched cancers [Cancer Genome Atlas Network, 2012].
Loss of Rb function in stem or progenitor cells is often a key event in neoplastic transformation [Sage, 2012] and is accompanied by epithelial–mesenchymal transition [Arima et al. 2012]. According to TCGA data, loss of Rb due to RB1 gene deletion occurs overall in 2–4% of breast cancers, and loss due to RB1 truncating mutations in 1–3% [Johnson et al. 2016], depending on breast cancer subtype, with up to 20% mutation/loss rate in basal-like tumors [Cancer Genome Atlas Network, 2012]. This leads to constitutive activation of E2F and induction of cyclin E and CDK2, independently from cyclin D-CDK4/6 activation, and is frequently accompanied by upregulation of p16 [Subhawong et al. 2009] due to a feedback loop [Kotake et al. 2007]. Other authors report higher frequencies of Rb loss of heterozygosity, correlating with low mRNA expression, particularly in triple-negative but also in luminal B breast cancers [Herschkowitz et al. 2008]. Rb functional inactivation may result also from cyclin D1 overexpression or p16INK4A inactivation, and is frequent in breast cancer, as shown by studies of Rb-loss gene signatures identifying tumors with deregulated Rb [Ertel et al. 2010; Herschkowitz et al. 2008]. Immunohistochemical assessment confirms more frequent Rb loss in triple-negative than in other breast cancer subtypes, and highlights its relation with benefit from adjuvant chemotherapy [Treré et al. 2009].
CDKN2A is deleted in 3–8% of breast cancers, more frequently in the triple-negative subtype [Johnson et al. 2016; Cairns et al. 1995], and may be mutated or silenced by promoter methylation [Ruas and Peters, 1998].
E2F transcription factors are not frequently mutated, but may be involved large-scale chromosomal aberrations such as loss of 16q [Johnson et al. 2016].
p21Cip1 expression is frequently reduced as a consequence of TP53 mutation [Musgrove et al. 1995] or Myc overexpression [Mukherjee and Conrad, 2005], and p27Kip1 expression is reduced as a result of HER2 amplification [Chu et al. 2008].
In summary [Cancer Genome Atlas Network, 2012; Witkiewicz and Knudsen, 2014], in luminal breast cancers, ER signaling induces cyclin D1 transcription, and there is often, particularly in luminal B tumors, cyclin D1 overexpression or gene amplification and CDK4 gain; p16INK4A or Rb losses are quite rare in luminal A, and somewhat more frequent in luminal B tumors. Triple-negative cancers have the highest rate of Rb inactivation and of p16INK4A overexpression, and may present cyclin E1 amplification. In HER2-positive tumors, cyclin D1 is often amplified or activated as a result of HER2 mitogenic signaling and there may be CDK4 gain. Aberrations of p16INK4A and those of Rb are mutually exclusive, as tumors with loss of p16INK4A have wild-type Rb, while tumors with mutant Rb show high expression of p16INK4A [Witkiewicz and Knudsen, 2014]. Within this framework, luminal tumors and, to some extent, HER2-positive tumors that often have an intact or overactive Rb pathway are those more likely to benefit from CDK4/6 inhibition.
Cell cycle and endocrine resistance
Estrogens stimulate cellular proliferation mainly by binding to nuclear ER alpha (ERα) that induces the expression of cyclin D1 and ultimately leads to Rb inactivation [Altucci et al. 1996]. They also inhibit the expression of CIP/KIP proteins and induce the expression of phosphatase Cdc25A [Foster et al. 2001], which further contribute to sustain cell proliferation. With a positive feedback, cyclin D1 can directly activate ERα [Zwijsen et al. 1997]. Although other mechanisms are implicated [Nardone et al. 2015], the cyclin D1-CDK4/6-Rb-E2F axis is critical for estrogen action.
Both selective ER modulators (SERMs) such as tamoxifen [Ichikawa et al. 2008] and selective ER downregulators (SERD) like fulvestrant [Carroll et al. 2000] lead to cell cycle arrest (CCA) in G1, reducing the expression of cyclins D and B and increasing that of p21Cip1, with fulvestrant also inducing accumulation of p130–E2F4 complexes characteristic of quiescence.
Aberrant expression or function of molecules involved in cell cycle regulation and in estrogen action have been implicated in endocrine resistance. Some examples are: the suppression or inhibition of CIP/KIP proteins [Chu et al. 2008; Pérez-Tenorio et al. 2006], which sometimes result from Myc [Mukherjee and Conrad, 2005] or cyclin D1 overexpression [Hui et al. 2002; Stendahl et al. 2004]; Rb inactivation [Bosco et al. 2007; Thangavel et al. 2011]; upregulation of CDK6, subtending resistance to fulvestrant [Alves et al. 2016]; hyperactivation of receptor tyrosine kinases, such as the epidermal growth factor receptor (EGFR) [McClelland et al. 2001], the human epidermal growth factor receptor 2 (HER2) [Knowlden et al. 2003], or the insulin-like growth factor receptor [Miller et al. 2009], that induce phosphorylation and activation of ERα or its coregulators; alterations of components of their downstream signaling pathways, such as MAPK/ERK and phosphoinositide 3-kinase (PI3K)/Akt [Miller et al. 2010]; ligand-independent, ERα-dependent activation of CDK4–Rb–E2F axis in estrogen-deprived breast cancer [Miller et al. 2011]; ERα gene (ESR1) gain-of-function mutations leading to constitutive receptor activation [Jeselsohn et al. 2015] that ultimately acts via CDK4/6–Rb [Luo et al. 2016].
Cyclin-dependent kinase 4/6 inhibitors: preclinical activity as single agents and mechanisms of response and resistance
First generation pan-CDK inhibitors showed modest clinical activity and considerable toxicity. More recently, selective small molecule CDK inhibitors have been developed, and CDK4/6 inhibitors are those in more advanced stage of development, with three compounds that have reached the clinical stage: palbociclib, ribociclib, and abemaciclib. Palbociclib is the one in more advanced development.
Palbociclib (PD 0332991) is an orally available pyridopyrimidine derivative, inhibiting CDK4/6 by binding to their ATP pockets with high selectivity, showing IC50 values for CDK4/cyclin D1, CDK4/cyclin D3, and CDK6/cyclin D2 of 11, 9, and 15 nmol/l, respectively, with low or absent activity against other kinases [Fry et al. 2004]. It potently inhibits cell proliferation, preventing progression of the cell cycle from G1 into the S phase, in Rb-positive cells of different tumor types, producing Rb dephosphorylation at specific serine residues as its pharmacodynamic hallmark.
Among human breast cancer cell lines representative of the different breast cancer subtypes, the ER-positive, luminal ones were the most sensitive, along with some HER2-amplified cell lines with luminal features, whereas cell lines with basal features were the most resistant [Finn et al. 2009]. Both endocrine-sensitive and endocrine-resistant lines may respond to CDK4/6 inhibitors [Petrossian et al. 2016]. High levels of cyclin D1 and of Rb, and low levels of p16 were predictors of sensitivity to palbociclib [Finn et al. 2009]. This was confirmed also in ex vivo studies on primary human tumor cultures [Dean et al. 2012b]. CDK4 mutations [Young et al. 2014], CDKN2A and CDKN2C (encoding p18INK4C) deletions [Wiedemeyer et al. 2010] and low E2F expression [Logan et al. 2013] have been shown to predict palbociclib response in other tumor types, whereas high expression of cyclin E1 [Konecny et al. 2011] was associated with resistance.
Breast cancer cell lines with an inactive cyclin D–CDK4/6–Rb pathway, usually because of loss of Rb and consequent upregulation of p16INK4A and downregulation of cyclin D1, are resistant to palbociclib [Dean et al. 2010]. This typically occurs in basal cell lines. Rb-proficient cells may become temporarily resistant to palbociclib after prolonged exposure that leads to increased p107 and CDK2 expression with or without loss of p21Cip1 and p27Kip1, but usually remain sensitive to deferred second rounds of treatment. Loss of Rb function would instead lead to true, long-term resistance, due to the increased transcription of cyclin A and E, which activates CDK2 and can drive the cell cycle independently of CDK4/6. Rb function is necessary for the induction of senescence by palbociclib, wherein tumor cells permanently exit the cell cycle [Dean et al. 2010].
Rb-knockdown experiments in breast cancer cell lines confirm that Rb status plays a prominent role in acute response to palbociclib, but shows partial activity of palbociclib also in some Rb-knocked-down cell lines, due to a compensatory role of p107, which is dephosphorylated in response to palbociclib, leading to E2F inhibition [Dean et al. 2010]. Some activity was seen also in other Rb-deficient cells owing to this mechanism [Rivadeneira et al. 2010].
Rarely, Rb-positive cell lines are resistant to palbociclib, and Rb phosphorylation does not decrease after drug exposure [Finn et al. 2009]. CKI downregulation and CDK2 reactivation, replacing CDK4/6, has been demonstrated in such cases in models of acute myelogenous leukemia [Wang et al. 2007]. Recently, ER-positive breast cancer cell lines have been shown to develop early adaptation to CDK4/6 inhibitors through cyclin D1–CDK2 complexes mediating G1–S transition; this ultimately results in acquired resistance to CDK4/6 inhibitors through selection of cells with Rb loss or CCNE1 amplification [Herrera-Abreu et al. 2016].
Recent studies on breast cancer cell lines have highlighted different features of clones with acquired palbociclib resistance: genomic deletion of RB1 in some cases, and Rb retention in others, with upregulation of E2F, TGFβ, Wnt, or NF-kB pathways [Lee et al. 2016]; increased activity of the PI3K/AKT/mTOR and changes in p53, apoptotic regulation and Rho/Rac pathway [Lenihan et al. 2016]; increased levels of 3-phosphoinositide-dependent protein kinase 1 (PDK1) [Jansen et al. 2016].
Activity of palbociclib has also been shown in some triple-negative breast cancer cell lines, belonging to the luminal androgen receptor (LAR) and the mesenchymal stem-like (MSL) subtypes [Asghar et al. 2015]. Sensitivity was associated with expression of androgen receptor and with low expression of cyclin E1.
In mouse xenograft models, palbociclib showed significant antitumor activity in breast cancer and in other tumor xenografts, inducing tumor regression in vivo, including some durable complete remissions, despite its cytostatic effect in vitro [Fry et al. 2004]. Palbociclib has also been shown to inhibit breast cancer cell migration and invasion, epithelial–mesenchymal transition, and metastatization, via inhibition of the c-Jun/COX-2 pathway [Qin et al. 2015].
Cyclin-dependent kinase 4/6 inhibitors: preclinical activity in combination
The combination of palbociclib and tamoxifen showed synergism in ER-positive breast cancer cell lines, likely due to the concomitant cyclin D1 inhibition by tamoxifen and CDK4/6 inhibition by palbociclib. Furthermore, palbociclib monotherapy was active in tamoxifen-resistant MCF7 cell lines, and partially restored sensitivity to tamoxifen in resistant lines [Finn et al. 2009].
Palbociclib has also been shown to resensitize fulvestrant-resistant cells to fulvestrant [Alves et al. 2016].
Estrogen deprivation, occurring during therapy with aromatase inhibitors (AIs), leads to acquired resistance through both ligand-independent, ERα-dependent activation of the CDK4-Rb-E2F axis [Miller et al. 2011] and PI3K hyperactivation [Miller et al. 2010], whereas resistance to PI3K inhibitors also involves activation of the cyclin D-CDK4-Rb-E2F axis [Vora et al. 2014]. CDK4/6 inhibitors have shown activity in AI-resistant and long-term estrogen-deprived (LTED) cell lines [Petrossian et al. 2016]. In mouse xenograft models of LTED breast cancer, the combination of fulvestrant with the pan-PI3K inhibitor BKM120 (buparlisib) was synergic [Miller et al. 2011], while in PI3K inhibitors’ resistant, PIK3CA-mutant breast cancer xenografts, synergism was demonstrated between PI3K inhibitors (the pan-class I inhibitor pictilisib or the PI3K-α specific inhibitor alpelisib) and the CDK4/6 inhibitor ribociclib [Vora et al. 2014].
Combined treatment with CDK4/6 inhibitors plus an endocrine agent or a PI3K inhibitor prevented cell adaptation to CDK4/6 inhibitors, and a triple combination of an endocrine agent plus a CDK4/6 and a PI3K inhibitor was more effective than paired combinations both in vitro, triggering apoptosis, and in patient-derived tumor xenografts [Herrera-Abreu et al. 2016]. In cases of acquired resistance due to CCNE1 amplification, treatment with a CDK2-inhibitor was able to resensitize tumor cells to CDK4/6 inhibitors. PI3K inhibition was synergistic with CDK4/6 inhibition, also in PIK3CA-mutated LAR and MSL triple-negative cell lines [Asghar et al. 2015].
Palbociclib, in combination with a new class of SERM/SERD hybrids, has shown activity in endocrine-resistant breast cancer cell lines and in ER gene (ESR1)-mutant cell lines and patient-derived tumor xenografts [Wardell et al. 2015; Nguyen et al. 2016].
Also ribociclib has shown single-agent activity in xenograft models of ER-positive breast cancer that was increased by the addition of letrozole or fulvestrant and by the PI3K inhibitors buparlisib and alpelisib [O’Brien et al. 2014].
Palbociclib also showed synergism with trastuzumab in HER2-amplified cell lines [Finn et al. 2009]. Tumors progressing after anti-HER2 therapy often show cyclin D1 overexpression, and abemaciclib has shown activity against anti-HER2-resistant tumors in mouse models, synergizing with anti-HER2 therapies and restoring sensitivity to anti-HER2 drugs [Goel et al. 2016]. Cyclin D1–CDK4 inactivates the tuberous sclerosis complex 2 (TSC2) by phosphorylation, relieving its inhibitory activity on mTORC1; mTORC1 can then exert its feedback inhibition of upstream HER receptors [Chandarlapaty et al. 2011]. Inhibition of CDK4/6 then restores HER receptors’ activity, resensitizing tumors to HER2 blockade [Goel et al. 2016].
The combination of palbociclib with the mTOR inhibitor everolimus yielded significant activity in preclinical models of non-small cell lung cancer [Gopalan et al. 2013]. Preclinical evidence of effectiveness of the combination of CDK4/6 inhibitors with inhibitors of the MAPK pathway also exists, particularly in melanoma [Yadav et al. 2014] and in colorectal cancer [Ziemke et al. 2016]. Additionally, the PDK1 inhibitor GSK2334470 was synergistic with CDK4/6 inhibitors in breast cancer cell lines and in xenograft models and was able to restore sensitivity to CDK4/6 inhibitors [Jansen et al. 2016].
The association of palbociclib with chemotherapeutic agents has often yielded antagonism, particularly in Rb-proficient cell lines [Roberts et al. 2012; McClendon et al. 2012]. Palbociclib did not resensitize paclitaxel-resistant cells to paclitaxel-induced apoptosis [Trapé et al. 2016]. Nonetheless, a short exposure to palbociclib to synchronize cells prior to paclitaxel resulted in increased cytotoxicity [Dean et al. 2012a]. The combination of concurrent palbociclib and radiation, or radiation followed by palbociclib (but not the reverse sequence), outperformed each single treatment modality in glioblastoma xenograft models, inhibiting DNA double-strand-break repair and promoting apoptosis [Hashizume et al. 2016].
Palbociclib clinical development
Phase I dose-finding and pharmacokinetics studies
The main phase I studies of palbociclib, enrolling patients with Rb-positive solid tumors or non-Hodgkin’s lymphoma, explored two different schedules: daily treatment for 2 weeks, followed by 1 week off treatment (schedule 2/1) [Schwartz et al. 2011] and daily treatment for 3 weeks, followed by 1 week off (schedule 3/1) [Flaherty et al. 2012]. The recommended phase II doses were 200 mg daily and 125 mg daily, respectively. The dose-limiting toxicities were hematological (neutropenia and thrombocytopenia) with both schedules, and the most common nonhematological adverse events were mild or moderate fatigue, nausea, diarrhea and constipation. No clinically significant effects on QTc interval were reported. Some clues of activity were evident, also in patients with breast cancer.
Palbociclib showed a mean absolute bioavailability after a dose of 125 mg of 46%, slightly increased by food; the pharmacokinetic was linear within the clinically relevant dose range, with a mean terminal half-life of about 26 hours, and steady state was reached within 8 days. Metabolism is mainly hepatic through cytochrome P450 3A (CYP3A) and sulfotransferase 2A1 (SULT2A1), and palbociclib is a weak CYP3A inhibitor. According to a population pharmacokinetic analysis, no dose adjustment is required for patients with mild or moderate renal impairment or with mild hepatic impairment [Sun and Wang, 2014]. A physiologically based pharmacokinetic model [Yu et al. 2016] predicts negligible effect of weak CYP3A inhibitors, while moderate CYP3A inhibitors may increase plasma area under the curve (AUC) by ~40% and moderate CYP3A inducers may reduce plasma AUC by ~40%.
The schedule 3/1 at a dose of 125 mg daily was selected for clinical development.
Palbociclib is a substrate of both P-glycoprotein and breast cancer resistance protein, which limits its brain distribution in preclinical models [de Gooijer et al. 2015]. Abemaciclib, on the contrary, has been shown to cross the blood–brain barrier [Patnaik et al. 2016].
Phase Ib/II studies
A single arm, phase II study of palbociclib monotherapy (schedule 3/1) enrolled 37 patients with Rb-positive (assessed by immunohistochemistry) advanced breast cancer [DeMichele et al. 2015]: 33 with hormone receptor (HR)-positive (HER2-positive in two cases) and four with triple-negative tumors. A clinical benefit (objective response or stable disease ⩾ 6 months) was achieved in 7 of 33 HR-positive patients (21%) and in none of the triple negatives (whose enrollment was stopped early), and progression-free survival (PFS) was 4.5 months and 1.5 months in the two subgroups, respectively. The two HER2-positive, HR-positive patients yielded a partial response and a stable disease lasting 5 months. The drug confirmed a good tolerability, and none of the biomarkers assessed (immunohistochemical staining for Rb, Ki67 and p16, and CCND1 amplification by FISH) correlated with clinical benefit.
Given the key roles of the estrogen and CDK4/6 pathways as drivers of breast cancer, and the preclinical evidence of activity in luminal tumors and of synergism with endocrine agents, clinical trials initially explored the combination of palbociclib with endocrine agents.
A small phase Ib study in postmenopausal patients with HR-positive, HER2-negative advanced breast cancer showed a good tolerability of the combination of palbociclib 125 mg daily (schedule 3/1) and letrozole 2.5 mg daily continuously, with no pharmacokinetic interactions [Slamon et al. 2010].
An open label, international, randomized phase II study assessed the efficacy and safety of palbociclib plus letrozole in postmenopausal patients with ER-positive, HER2-negative breast cancer, as first-line therapy for advanced disease (PALOMA-1/TRIO-18 trial) [Finn et al. 2015]. Patients could have received neoadjuvant or adjuvant endocrine treatment, but, if this included letrozole, it would need to have been stopped at least 1 year before study entry. Two separate cohorts were enrolled sequentially: cohort 1 included any patient with ER-positive, HER2-negative disease, while in cohort 2, further requirements were either amplification of CCND1, or loss of CDKN2A, or both, assessed at a central laboratory. Patients were randomly allocated 1:1 to receive either palbociclib 125 mg daily for 3 weeks of any 4-week cycle, plus letrozole 2.5 mg daily continuously, or letrozole 2.5 mg daily alone. The intention of cohort 1 was to provide preliminary safety and efficacy data, and the analysis of the primary endpoint of PFS was initially intended as based on cohort 2 only, after enrollment of 150 patients. However, an unplanned interim analysis of cohort 1 showed relevant activity of palbociclib, with PFS hazard ratio (HR) of 0.35 (95% confidence interval (CI), 0.17–0.72; p = 0.006) favoring the combination arm. This preliminary analysis also suggested that further patient selection based on biomarkers was unlikely to further increase the difference between treatment arms. Therefore, cohort 2 was closed in advance, and the study was amended, planning a combined analysis of cohorts 1 and 2 for the primary endpoint of PFS. After enrollment of 165 women, stratified by disease site and disease-free interval, 84 were randomized to palbociclib plus letrozole and 81 to letrozole alone, and after almost 30 months (median) follow up, the median PFS was 20.2 months for the combination arm versus 10.2 months for letrozole alone, with HR, 0.488; 95% CI, 0.319–0.748; p = 0.0004. The difference in PFS between the two arms was significant in both cohorts 1 and 2, and was consistent across patient subgroups defined by demographic, clinical and biological features [Finn et al. 2016a], apart from the lack of significant difference between the study arms in the subgroup of patients with disease recurrence within 12 months from the end of adjuvant therapy. Also, the rates of objective responses (ORR, 43% versus 33%; p = 0.13) and clinical benefit (CBR, 81% versus 58%; p = 0.0009) were higher in the combination arm, whereas overall survival (OS) was not different between the two arms (HR, 0.813; 95% CI, 0.492–1.345; p = 0.42). Treatment was well tolerated and the mean relative dose intensity for palbociclib was 94%. Leukopenia, neutropenia and fatigue were the most common adverse events in the palbociclib arm. An evaluation of pain by means of the Brief Pain Inventory showed no significant differences in pain severity and interference with daily activities between the two arms [Bell et al. 2016].
On the basis of these results, in February 2015, the US Food and Drug Administration (FDA) granted accelerated approval to palbociclib for use in combination with letrozole for the treatment of postmenopausal women with ER-positive, HER2-negative advanced breast cancer, as initial endocrine-based therapy for their metastatic disease [US Food and Drug Administration, 2015].
Based on preclinical data of palbociclib synergism with paclitaxel when administered on an alternating schedule, enabling tumor cell synchronization before the administration of paclitaxel, a phase Ib trial, including a dose escalation and an expansion cohort, identified palbociclib 75 mg daily as optimal combination for 4 days of run-in, and then on days 2–4, 9–11, 16–18 of each 28-day cycle, plus paclitaxel 80 mg/m2 weekly for three cycles and then on days 1, 8, 15 of 28-day cycles [Clark et al. 2016]. Dose-limiting toxicities were grade 3 transaminitis and febrile neutropenia, and uncomplicated grade 3 and 4 neutropenia was the most common adverse event. Among 24 evaluable patients, 14 (58%) had a partial response or stable disease longer than 6 months, supporting further development of the regimen.
Phase III studies
In the double blind, phase III PALOMA-3 trial, patients with HR-positive, HER2-negative advanced breast cancer that had relapsed or progressed during prior endocrine therapy (within 12 months from completion of adjuvant endocrine therapy, or during or within 1 month after the end of prior endocrine therapy for advanced disease) with tamoxifen if pre- or perimenopausal, or with an AI if postmenopausal, were randomized in a 2:1 ratio to palbociclib (125 mg daily for 3 weeks, followed by 1 week off over 4-week cycles) plus fulvestrant (500 mg intramuscularly on days 1, 15, 29, then every 4 weeks) or to placebo plus fulvestrant [Turner et al. 2015; Cristofanilli et al. 2016]. Pre- and perimenopausal patients received also goserelin 3.6 mg subcutaneously every 4 weeks, starting at least 4 weeks before randomization. One prior line of chemotherapy for advanced disease was allowed. Of 521 patients enrolled, stratified by disease site, menopausal status, and sensitivity to prior endocrine therapy, 347 received palbociclib plus fulvestrant and 174, placebo plus fulvestrant. Almost 80% of the patients had tumors that had been sensitive to prior endocrine therapy. At a median follow up of 8.9 months, median PFS was 9.5 months for fulvestrant plus palbociclib versus 4.6 months for fulvestrant plus placebo, with HR, 0.46 (95% CI, 0.36–0.59; p < 0.0001). The ORR in patients with measurable disease was 25% in the palbociclib arm and 11% in the placebo arm (p = 0.0012), with median time to response of 112 days and 57 days respectively, and the CBR was 64% and 36%, respectively (p < 0.0001). Results from subgroup analyses were generally consistent with those from the overall population. In particular, the degree of endocrine sensitivity (either in terms of previous endocrine response or HR expression) did not affect benefit, nor did the menopausal status [Loibl et al. 2016]. Although PIK3CA mutations (evaluated on circulating cell-free DNA on 394 patients, with a mutation rate of 33%) conferred a nonsignificant worsening in PFS, the magnitude of benefit from palbociclib was independent from PIK3CA status. Likewise, while ESR1 gene mutations conferred a marginally significant worsening of PFS compared with wild-type status, benefit from palbociclib was independent from ESR1 status [Fribbens et al. 2016]. Grade 3 or 4 neutropenia occurred in 65% of patients receiving palbociclib versus 1% of those receiving placebo; nonetheless, febrile neutropenia was uncommon (1%) in both groups. Adverse events with >10% increased frequency in the palbociclib group were neutropenia, leucopenia, anemia, thrombocytopenia, stomatitis, alopecia, rash, infections and fatigue; these were mainly mild or moderate, and treatment discontinuation due to adverse events occurred in 4% in the palbociclib group and 2% with placebo [Verma et al. 2016]. The median time to onset of grade ⩾3 neutropenia was 16 days, and the median duration of grade ⩾3 neutropenia, 7 days. The mean relative dose intensity for palbociclib plus fulvestrant was 86%, but dose reductions due to neutropenia did not have a detrimental effect on efficacy. The combined treatment significantly delayed deterioration of global quality of life and improved pain compared with the placebo arm [Harbeck et al. 2016].
On the basis of PALOMA-3 results, on 19 February 2016, the FDA approved palbociclib for use in combination with fulvestrant for the treatment of women with HR-positive, HER2-negative advanced or metastatic breast cancer with disease progression following endocrine therapy [US Food and Drug Administration, 2016].
Results of the double blind, phase III PALOMA-2 trial, designed to confirm those from PALOMA-1, were presented at the 2016 ASCO meeting [Finn et al. 2016b]. In the trial, 666 postmenopausal patients with HR-positive, HER2-negative advanced breast cancer, not previously treated for advanced disease, stratified for disease site, disease-free interval, and prior (neo)adjuvant hormonal therapy, were randomized 2:1 to palbociclib plus letrozole (same doses and schedule as in PALOMA-1) or placebo plus letrozole. Baseline characteristics were well balanced among groups. After a median follow up of about 23 months, median PFS was 24.8 months in the palbociclib arm and 14.5 months in the placebo arm, with HR 0.58 (95% CI, 0.46–0.72; p < 0.000001). The ORR was 42% in the palbociclib arm versus 35% with placebo (p = 0.031) and the CBR was 85% and 70%, respectively (p < 0.0001). Benefit from palbociclib was apparent across all prespecified subgroups. The median relative dose intensity for palbociclib was 93%. Common adverse events (any grade, palbociclib versus placebo group) were: neutropenia (79.5% versus 6.3%), fatigue (37.4% versus 27.5%), nausea (35.1% versus 26.1%), arthralgia (33.3% versus 33.8%) and alopecia (32.9% versus 15.8%). Febrile neutropenia was seen only with palbociclib (2.5%). Permanent discontinuation due to AEs was 9.7% with palbociclib versus 5.9% with placebo. The OS analysis is pending.
The low incidence of febrile neutropenia compared with the high incidence of grade 3/4 neutropenia is due to the cytostatic effect of palbociclib on bone marrow cells that is reversible upon drug withdrawal, contrary to chemotherapeutic agents that induce bone marrow progenitors’ apoptosis [Hu et al. 2016].
Neoadjuvant studies
A phase II neoadjuvant study has been conducted in patients with clinical stage II/III ER-positive, HER2-negative breast cancer [Ma et al. 2016] to assess molecular changes induced by anastrozole and palbociclib. Therapy consisted of anastrozole alone for 28 days, followed by anastrozole plus palbociclib (schedule 3/1) for four 28-day cycles, followed by 2–4 weeks with anastrozole alone before surgery. A subgroup of patients also received palbociclib for 10–12 days immediately prior to surgery. The primary endpoint was the rate of complete CCA, defined as Ki67 ⩽ 2.7%, at an early biopsy taken on day 15 of the first cycle of combined treatment. Premenopausal patients also received goserelin. Preliminary results were reported at the 2015 San Antonio Breast Cancer Symposium: 39 of 45 evaluable patients achieved CCA (87%), including 22 of 28 patients (79%) with wild-type PIK3CA, 15 out of 15 patients with mutant PIK3CA and 2 out of 2 with undetermined PIK3CA. Among 43 patients who also had a biopsy just before starting palbociclib, anastrozole alone induced CCA in 11 cases (26%), whereas 15 days of palbociclib converted non-CCA to CCA in another 26 patients (60%). The combination was ineffective in two patients with nonluminal cancers and in a subset of luminal B cancers. Clinical responses were seen in 67% of the patients and no pathological complete response was seen. While patients who stopped palbociclib 2–4 weeks before surgery showed a rebound in Ki67 assessed at surgery, those who received palbociclib immediately before surgery did not, highlighting the cytostatic action of the drug, more apt for maintenance treatment.
In a further short-term preoperative trial, aimed at assessing the CCA rate and to identify predictive biomarkers, 100 patients with untreated breast cancer were randomized 3:1 to palbociclib 125 mg daily for 14 days until the day before surgery, versus no treatment. The CCA rate was 58% with palbociclib versus 10% in the control arm. No Ki67 response was observed in triple-negative and in HER2-positive tumors. While baseline Rb, phosphorylated Rb and p16 did not predict response, responses were associated with changes in phosphorylated Rb from baseline [Arnedos et al. 2016].
Studies with other cyclin-dependent kinase 4/6 inhibitors
Several clinical trials are ongoing with the other CDK4/6 inhibitors ribociclib and abemaciclib, and have been reviewed elsewhere [O’Leary et al. 2016; Barroso-Sousa et al. 2016]. Particularly relevant are studies of triple drug combinations. Preliminary results have been reported for a phase Ib/II study of triplet therapy with ribociclib, everolimus, and exemestane in 77 evaluable postmenopausal women with HR-positive, HER2-negative advanced breast cancer [Bardia et al. 2016], demonstrating feasibility and a disease control rate (patients who don’t have disease progression as best response) of 73% in heavily pretreated patients. In a further phase Ib/II study, 46 patients received a combination of ribociclib, alpelisib and letrozole that showed an acceptable safety profile and a disease control rate of 70% [Juric et al. 2016].
Ongoing clinical trials with palbociclib
Several phase Ib, II, and III studies are ongoing with palbociclib (Table 1). Among these are: some adjuvant studies, such as the phase III PALLAS trial comparing endocrine therapy of physician choice for at least 5 years with endocrine therapy for at least 5 years plus palbociclib for 2 years, and the PENELOPE-B trial, with standard endocrine treatment plus palbociclib or placebo in patients with residual disease after neoadjuvant chemotherapy; several neoadjuvant studies, both in HER2-positive, HR-positive and in HER2-negative, HR-positive breast cancer, some of which comparing endocrine therapy plus palbociclib with chemotherapy; several trials in the metastatic setting, either comparing endocrine therapy plus palbociclib with chemotherapy, or exploring combinations of palbociclib with other new drugs such as new endocrine agents, PI3K inhibitors (the pan-class I PI3K inhibitor pictilisib and the selective inhibitor of class I PI3K α, γ, and δ isoforms, taselisib), anti-PD-1 monoclonal antibodies and anti-HER2 drugs. Studies of triple combinations of palbociclib with an endocrine agent (fulvestrant or letrozole) and the PI3K/mTOR inhibitor gedatolisib are also ongoing.
Ongoing interventional trials with palbociclib in breast cancer.
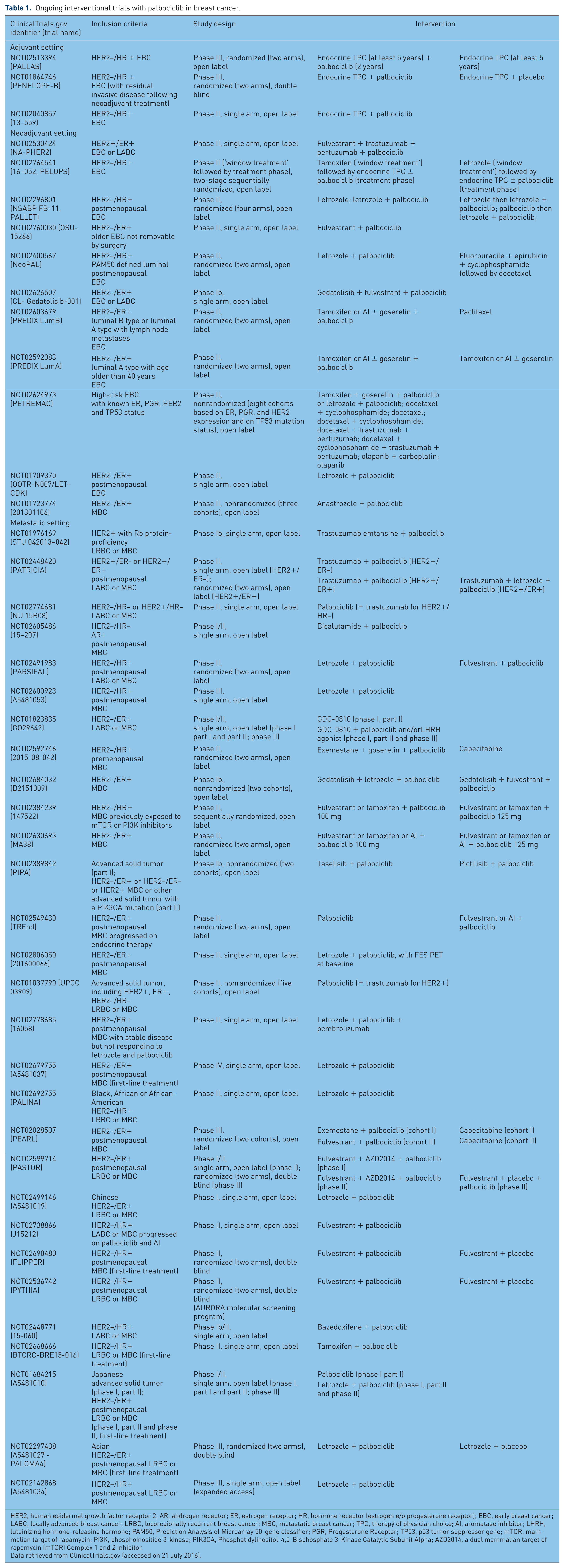
HER2, human epidermal growth factor receptor 2; AR, androgen receptor; ER, estrogen receptor; HR, hormone receptor (estrogen e/o progesterone receptor); EBC, early breast cancer; LABC, locally advanced breast cancer; LRBC, locoregionally recurrent breast cancer; MBC, metastatic breast cancer; TPC, therapy of physician choice; AI, aromatase inhibitor; LHRH, luteinizing hormone-releasing hormone; PAM50, Prediction Analysis of Microarray 50-gene classifier; PGR, Progesterone Receptor; TP53, p53 tumor suppressor gene; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinase; PIK3CA, Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha; AZD2014, a dual mammalian target of rapamycin (mTOR) Complex 1 and 2 inhibitor.
Data retrieved from ClinicalTrials.gov (accessed on 21 July 2016).
Open questions and future directions
Palbociclib received FDA-accelerated approval in HER2-negative luminal breast cancer, both as initial endocrine-based therapy for metastatic disease, in combination with letrozole, and after progression, following an endocrine therapy for advanced disease, in combination with fulvestrant. The best positioning of palbociclib in the therapeutic strategy of luminal tumors remains an open question, partly intertwining with the still-open issue of the best sequencing of endocrine agents in the metastatic setting. Although the degree of endocrine sensitivity does not seem to affect the benefit from palbociclib, some distinctions are worthy. While more aggressive tumors certainly deserve the use of palbociclib from the first line of treatment, some indolent or highly endocrine-sensitive tumors may also have long PFS with endocrine therapy alone. Therefore, the trade-off between improved efficacy and added side effects may differ, depending on patient and tumor characteristics and line of treatment. On the other hand, everolimus, another agent that improves the clinical outcome when added to endocrine therapy, was studied in patients progressing on, or shortly after, therapy with a nonsteroidal AI, and will more likely, but not necessarily, be used after therapy with palbociclib, although the efficacy of everolimus plus exemestane after progression to CDK4/6 inhibitors (or vice versa) has not been addressed in clinical trials. A further open issue that is being addressed in clinical trials is the efficacy of continuing CDK4/6 inhibition beyond progression to palbociclib, for example, maintaining palbociclib while changing the endocrine agent, or changing the CDK4/6 inhibitor.
The studies of triple blockade are very intriguing, combining an endocrine agent with a CDK4/6 inhibitor with another drug, like a PI3K, a MAPK, or an mTOR inhibitor, or an anti-HER2, ideally chosen based on tumor genomic profile. Should these drug combinations prove tolerable, they could have the potential to be not only cytostatic, but to induce tumor cell apoptosis, with greater potential activity. Very active targeted drug combinations would be worth exploring in the first-line setting, even as an alternative to chemotherapy in aggressive tumors, as well as in the neoadjuvant setting.
Balancing benefits with toxicity and costs will be critical in the adjuvant setting [Carey and Perou, 2015], should palbociclib demonstrate efficacy in this context, given that a relevant fraction of patients with luminal tumors would be cured with surgery alone. In this regard, studies in the neoadjuvant setting may help to identify predictors of response. The potential of 3-deoxy-3[(18)F]fluorothymidine (FLT) positron emission tomography in early response assessment has been explored in lymphomas [Leonard et al. 2012] and deserves study in breast cancer too, but further genomic and proteomic studies are likely needed for more accurate prediction.
Conclusion
Palbociclib is the first member of the CDK4/6 inhibitors entering the clinical arena. With 10-month improvement in PFS in first-line metastatic HR-positive, HER2-negative breast cancer, when added to letrozole, it represents one of the best steps forward in the treatment of luminal breast cancer. Nonetheless, an impact on OS is still unproven, given the limited follow up of the phase III trials available. It also shows promise in other breast cancer subtypes, especially in a subset of HER2-positive tumors. Its single-agent activity is basically cytostatic, and combination with other agents is usually required to induce tumor cell senescence or apoptosis. Identifying response predictors will be essential for a rational use of the drug, to avoid unnecessary toxicity and costs [Matter-Walstra et al. 2016]. Given the complexity of the pathways regulating cell cycle, no single biomarker has emerged as predictor of response, beyond the presence of HRs. Further, deeper efforts in the characterization of a single tumor’s biomarkers are ongoing, and a systemic approach will likely be necessary to better identify biomarkers’ constellations associated with response or resistance, as well as the optimal combination therapies for any single tumor.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.