
Abstract
Human epidermal growth factor receptor 2 was the first successful molecular target in treating gastric cancer, marking a significant milestone for targeted therapies. Emerging evidence on Claudin18.2 (CLDN18.2) has recently reshaped the paradigm of therapeutic targets, expanding the focus beyond conventional oncogenic drivers. Therapeutic strategies now target tumor-associated molecules which highly expressed in tumors but are not necessarily critical for tumor growth or survival. Molecules such as trophoblast cell surface antigen 2, Caprin-1, and Nectin-4 are promising non-oncogenic targets for advanced gastric cancer treatment. Innovative therapeutic approaches, such as antibody-drug conjugates, bispecific antibodies, and chimeric antigen receptor T-cell therapy, have accelerated the potential of targeting tissue-associated antigens. This review provides an update on CLDN18.2-directed therapies and explores the development of novel therapeutic strategies targeting non-oncogenic drivers. In addition, we discuss ongoing challenges, including biomarker overlap, resistance mechanisms, and future directions for next-generation molecular targeted therapy in gastric cancer.
Introduction
Despite the availability of several approved agents for unresectable advanced gastric cancer or gastroesophageal junction cancer (GC/GEJC), the prognosis remains poor.1,2 While targeted therapies have improved survival in select patients with GC/GEJC,1,2 their benefits remain limited to a subset of the population. Thus, identifying new therapeutic targets is urgently needed to advance treatment options for unresectable advanced GC/GEJC.
The concept of molecular targeted therapy began with the aim of exploiting cancer’s Achilles’ heel—selectively attacking vulnerabilities essential for cancer survival. Kinase inhibition became a key strategy, exemplified by imatinib, which revolutionized chronic myeloid leukemia 3 and gastrointestinal stromal tumor treatment 4 with improved outcomes and lower toxicity.
Targeted therapy initially focused on oncogenic drivers—genetic alterations that drive tumor growth. Human epidermal growth factor receptor 2 (HER2) was the first successful target, with trastuzumab improving survival in HER2-positive breast cancer. 5 This success led to HER2-targeted therapy in GC/GEJC, where the ToGA trial confirmed a survival benefit with trastuzumab plus chemotherapy. 6
However, subsequent attempts to target other receptor tyrosine kinases, such as epidermal growth factor receptor (EGFR,7–9 mesenchymal-epithelial transition factor (MET),10,11 and fibroblast growth factor receptor (FGFR), 12 have failed to show meaningful survival benefits. This limited efficacy can be attributed primarily to the molecular heterogeneity of GC/GEJC, which gives rise to intra-/inter-tumoral diversity and clonal evolution, compromising the effectiveness of targeted therapies. 13
In contrast to oncogenic drivers, non-oncogenic targets are molecules that do not directly causing tumorigenesis, but are involved in tumor proliferation, survival, or adaptation. These are often normal proteins with abnormal or overexpressed levels in cancer cells. Although no consensus on non-oncogenic targets has been established, vascular endothelial growth factor (VEGF) or programmed death-ligand 1 (PD-L1) meets the definition mentioned above. Ramucirumab, 14 a monoclonal antibody targeting VEGFR2, and immune checkpoint inhibitors such as nivolumab,15–17 pembrolizumab,18,19 and tislelizumab, 20 are already available in clinical practice, highlighting the potential of such targets to expand therapeutic options. CLDN18.2 has emerged as a new therapeutic target. As a key molecule involved in tight junction assembly in normal gastric epithelium, CLDN18.2 exhibits highly lineage-specific expression and ectopic expression in various cancers, underscoring its clinical significance. Zolbetuximab, a first-in-human chimeric IgG1 monoclonal antibody targeting CLDN18.2, demonstrated improved overall survival in patients with CLDN18.2-positive, HER2-negative GC/GEJC in the phase III SPOTLIGHT and GLOW trials, establishing a new standard of care.21,22 Following the success of CLDN18.2-directed therapy, non-oncogenic drivers, such as trophoblast cell surface antigen 2 (TROP2), Caprin-1, and Nectin-4, are targeted in treating GC/GEJC.
Previously, various reviews have focused on the role of VEGF and PD-L1 in treating GC/GEJC.23–25 This review will explore treatment approaches targeting other non-oncogenic drivers, focusing on CLDN18.2 and emerging targets in GC/GEJC. We will discuss their clinical significance, current advancements, and potential to transform the therapeutic landscape of this challenging disease.
Claudin18.2
Implications for carcinogenesis
Claudins are a family of 26 structurally related proteins in humans that constitute tight junctions in epithelial and endothelial tissues.26,27 Lineage specificity is a unique feature of CLDN18. It has two isoforms: CLDN18.2, which is highly specific to the normal gastric mucosa, and CLDN18.1, which is expressed in the lungs 28 (Figure 1). Tumor-specific ectopic expression of CLDN18.2 is a particularly intriguing aspect as a therapeutic target.29,30 Sahin and Türeci reported the ectopic expression of CLDN18.2 in pancreatic, esophageal, and lung cancers. 28 CLDN18.2 is located at the apical side of the paracellular space, where it forms tight junctions that maintain tissue-specific permeability and gastric epithelial cell polarity.31,32 CLDN18.2 protein expression is maintained during malignant transformation in approximately 70% of GC.29,33 Although upregulation of CLDN18 expression has been suggested to activate the ERK1/2 pathway, potentially promoting tumor cell proliferation, 34 definitive evidence supporting the role of CLDN18.2 in carcinogenesis is still lacking. 29 Meanwhile, the loss of CLDN18.2 has been investigated as a factor in malignant transformation. 35 In preclinical studies, knockdown of CLDN18 expression can immediately induce severe gastritis; however, multiple steps are required to develop gastric neoplasia in the mouse stomach, which takes a long time.32,35 Furthermore, activation of other oncogenic pathways, such as the Wnt signaling pathway, is necessary to initiate this process.
Claudin18.2 expression during malignant transformation. (a) In normal gastric epithelial cells, polarity is maintained, and Claudin18.2 is prominently expressed at the apical membrane within tight junctions. (b) As malignancy progresses, gastric epithelial cells lose their polarity due to EMT, disrupting Claudin18.2 expression. Downregulation of Claudin18.2 is frequently observed at the invasive front of tumors undergoing EMT.
Loss of CLDN18.2 expression reflects disruption of normal gastric function and indicates that cells have undergone malignant transformation, losing their original physiological roles. However, CLDN18.2 expression can still be retained in some gastric cancer cells during transformation. Conversely, loss of CLDN18.2 can also occur in non-malignant conditions such as intestinal metaplasia, where it is replaced by other claudin family proteins. This complexity underscores a key feature of non-oncogenic drivers—their context-dependent expression and roles in cancer.
The CLDN18.2-ARHGAP fusion is a structural aberration first reported in the comprehensive analysis of The Cancer Genome Atlas (TCGA) stomach. 36 Patients with CLDN18.2-ARHGAP fusions were more frequently observed in diffuse-type histology and early-onset GC/GEJC.37,38 The chemosensitivity and prognosis of patients with CLDN18-ARHGAP fusion-positive are poorer compared to fusion-negative, suggesting the aggressive nature of CLDN18.2-ARHGAP fusion-positive GC/GEJC.37,38 However, evidence on the impact of the CLDN18.2-ARHGAP fusion on cancer proliferation remains limited. Upon the formation of the fusion gene, CLDN18.2 loses its binding motif at the C-terminal, disrupting the actin-cytoskeleton-associated complex and reducing cell–extra cell matrix adhesion. 39 Sustained inhibition of RhoGAP by the CLDN18.2-ARHGAP fusion, anchored beneath the cell membrane, may maintain RhoA in its active state, thereby enabling anchorage-independent growth. 40 Preclinical research using diffuse-type cell lines harboring ARHGAP fusions revealed that ARHGAP-RhoA signaling disruption may contribute to resistance to anoikis, particularly through E-cadherin-mediated homotypic adhesion via the ROCK-MLC2 pathway. 41 Thus, ARHGAP fusions may share similar alterations in inherent signaling pathways with RHOA mutations. The expression level of CLDN18.2 is independent of CLDN18.2-ARHGAP fusion. 42 The function of the CLDN18-ARHGAP fusion is not yet fully understood. However, current evidence supports that CLDN18 merely serves as an anchor, providing a site for RhoGAP subcellular localization. The RhoGAP domain is critical in malignant transformation.43,44
Clinicopathological features
In the SPOTLIGHT and GLOW trials, the CLDN18.2 positivity rate, defined as ⩾75% of tumor cells with moderate to strong membranous staining intensity using Ventana OptiView CLDN18 (clone 43-14A), was 38.4%.21,22,45 Retrospective studies have shown that the overlap between CLDN18.2 positivity and HER2 positivity is approximately 4%, while the overlap with a PD-L1 combined positive score (CPS) of ⩾5 is 10%–27%. The overlap with microsatellite instability-high (MSI-H) status is rare, observed in only about 4% of cases. CLDN18.2-positive GC/GEJC is more common in younger patients, diffuse-type, type 4, and cases with peritoneal dissemination.42,45–47 These patients often do not benefit from preexisting molecularly targeted therapies or immune checkpoint inhibitors (ICIs). Studies also indicate that CLDN18.2 expression is not linked to prognosis.42,46,47
Zolbetuximab
Zolbetuximab, a first-in-class chimeric IgG1 monoclonal antibody targeting CLDN18.2, induces cancer cell death via antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity. Early-phase clinical trials of zolbetuximab demonstrated the utility of using CLDN18.2 immunohistochemistry (IHC) to enrich the study population. However, unlike trastuzumab in HER2-positive cancers, the efficacy of zolbetuximab is not associated with staining intensity (IHC 3+ vs 2+). 48 In the subsequent randomized phase II FAST trial, the additive effect of zolbetuximab was observed predominantly in patients with high CLDN18.2 expression (IHC 2+/3+ in ⩾70% of tumor cells), compared with those with moderate expression (2+/3+ in 40%–69%). 49 Based on these findings, later phase III trials restricted enrollment to patients with high CLDN18.2 expression. Two global phase III randomized trials (SPOTLIGHT and GLOW) independently demonstrated the efficacy and safety of zolbetuximab in combination with standard therapies.21,22 A combined analysis of these trials demonstrated significant improvements in progression-free survival (PFS) and overall survival (OS) with zolbetuximab plus chemotherapy compared to placebo plus chemotherapy. The median PFS was 9.2 months in the zolbetuximab group versus 8.2 months in the placebo group (HR 0.71; 95% CI, 0.61–0.83). For OS, the median was 16.4 months in the zolbetuximab group compared to 13.7 months in the placebo group (HR 0.77; 95% CI, 0.67–0.89). 50
Moreover, health-related quality of life assessments revealed that, due to the unique gastrointestinal (GI) toxicity of zolbetuximab, a transient deterioration occurred during the initial few cycles, which subsequently recovered in later cycles. Overall, the survival benefit appears to outweigh this temporary decline in quality of life. 51 However, further efforts to reduce GI toxicities will be essential for the implementation of anti-CLDN18.2 therapy into clinical practice.
Managing zolbetuximab-related gastrointestinal toxicities
Interaction with zolbetuximab and CLDN18.2 on normal stomach epithelium probably induces unique GI toxicities like nausea, vomiting, and decreased appetite. These GI toxicities were typically observed approximately 1 h after initiating zolbetuximab during the first cycle. These incidents showed a marked reduction in subsequent cycles. Temporary infusion interruptions often alleviated symptoms, allowing treatment to continue without long-term disruptions. 52 In the SPOTLIGHT and GLOW trials, GI toxicities were similar between the zolbetuximab and placebo groups in patients who underwent total gastrectomy. 53 Animal studies using ferrets revealed that zolbetuximab induces gastric mucosal injury soon after administration, with mucosal sloughing observed during vomiting and submucosal inflammation appearing 6 h after the onset of vomiting. In the study using ferrets, 54 combination therapy with antiemetic agents like 5-HT3, NK1, corticosteroid, and others was effective for mucosal injuries. Concerns about reduced ADCC activity led to the avoidance of steroids in 70% of SPOTLIGHT trials. However, post hoc analyses revealed that these were unfounded. 55 Excluding early discontinuations due to GI toxicities, the per-protocol population in SPOTLIGHT showed further improvements in PFS (HR 0.65) and OS (HR 0.69), with Kaplan–Meier curves separating early. These findings highlight the importance of early toxicity management in maintaining treatment continuity and maximizing efficacy. 55 According to Japanese guidelines, zolbetuximab-containing platinum doublet regimens are recommended to be managed as highly emetogenic chemotherapy. 56 The early administration of an additional antiemetic at the onset of mild nausea may help prevent its progression to more severe symptoms. 54 Step-up dosing is another key management for antiemesis induced by zolbetuximab. 52 Combination therapy with multiple antiemetic agents and step-up dosing could reduce the zolbetuximab-induced unique GI toxicities, resulting in 21% nausea and no vomiting in clinical experiences of daily practice. 57 Higher loading at the initiation of zolbetuximab might increase GI toxicities. A randomized phase II GENTLE-Z trial to assess the loading versus flat-dosing is under investigation in multicenters in Japan (jRCTs031240347). 58
Considerations for first-line treatment decisions in CLDN18.2-positive gastric cancer
Combination therapy, with either zolbetuximab or immune checkpoint inhibitor (ICI) (nivolumab or pembrolizumab), is a recommended regimen for HER2-negative, CLDN18.2-positive, unresectable, or recurrent GC/GEJC. A recent retrospective analysis revealed that nivolumab combined with chemotherapy showed consistent improvements in PFS and OS across CLDN18.2-positive and -negative cases (defined as moderate-to-strong expression in ⩾75% of tumor cells), even in subgroups stratified by PD-L1 CPS (⩾5 vs <5). 59 These findings suggest that CLDN18.2 expression status may not significantly impact ICI efficacy. Selecting the optimal regimen among competitive agents is a challenging issue in clinical practice.
The considerable efficacy of nivolumab or pembrolizumab for patients with MSI-H supports the preferential use of ICI with chemotherapy, even in CLDN18.2-positive cases. 60 Meanwhile, for patients with PD-L1 CPS <1, who are unlikely to benefit from ICIs,16–18 zolbetuximab appears more suitable. Furthermore, for patients with CPS ⩾10, HRs for death (0.65) in the subgroup of both CheckMate-649 and KEYNOTE-859 trials were consistently favorable compared with these (HR 0.75) of the SPOTLIGHT and GLOW trials,16,18,21,22 implying that more patients with CLDN18.2-positive and CPS ⩾10 are likely to benefit from the initial use of ICI. Although biomarkers will be useful for patient selection, microsatellite stable (MSS)/ proficient mismatch repair (pMMR), and PD-L1 CPS 1 ⩽ and <10 are the predominant subsets in patients with HER2-negative and CLDN18.2-positive GC/GEJC. 61 In addition, the availability of nivolumab as a later-line therapeutic option will influence the selection of the most reasonable treatment approach. Treatment selection should be individualized, considering each therapeutic approach’s distinct toxicity profiles and potential clinical benefits. The ILUSTRO study, a multi-cohort phase II trial, investigates new combination therapy possibilities for zolbetuximab. 62 Zolbetuximab in combination with mFOLFOX6 and nivolumab was evaluated in Cohort 4, while Cohort 5 focused on perioperative combination therapy with zolbetuximab and FLOT (NCT03505320). Effective management of GI toxicity is crucial for advancing zolbetuximab-containing therapy in the perioperative setting.
Newer monoclonal antibody targeting CLDN18.2
Osemitamab (TST001) is another monoclonal antibody targeting CLDN18.2 with a higher binding affinity than zolbetuximab, and preclinical data suggest stronger antitumor effects. The Transtar102 trial (NCT04495296), Cohort G, evaluates the efficacy and safety of osemitamab in combination with CAPOX and nivolumab 63 (Table 1). Patients with confirmed HER2-positive tumors were excluded, but those with varying CLDN18.2 and PD-L1 expression levels were eligible. CLDN18.2 expression was assessed centrally using the 14G11 LDT assay, categorizing patients into three groups: high/moderate (H/M), low (L), and negative (R), including two patients with unknown status. Data from 82 patients included 40 in the 3 mg/kg cohort and 42 in the 6 mg/kg cohort (including three in each Safety Run-in). Overall, 11.0% had GEJC (n = 9), 67.1% had intact stomachs (n = 55), 19.5% had peritoneal dissemination (n = 16), and 68.3% had PD-L1 CPS <5 (n = 56). Among 66 patients with evaluable CLDN18.2 and PD-L1 data, the ORR and mPFS were 58.1% and 12.6 months in the H/M group (n = 32), and 52.4%/7.1 months and 55.6%/8.5 months in the L (n = 22) and R (n = 28) groups, respectively. Treatment-related adverse events included nausea (67.1%, grade ⩾3: 3.7%), vomiting (59.8%, grade ⩾3: 2.4%), and decreased appetite (42.7%, grade ⩾3: 4.9%). Osemitamab shows potential efficacy even in patients with lower CLDN18.2 expression levels than those included in the zolbetuximab criteria. However, determining clear added benefits from the phase I/II results remains challenging, and confirmation in phase III trials is awaited.
Summary of phase I/II trials targeting non-oncogenic drivers for gastric/gastroesophageal junction cancer.

NCT04495296: IHC assay: 14G11, CLDN18.2 cutoff: Moderate–High: 2/3+ ⩾40%, Low: ⩾1+ in ⩾10% and ⩾2+ in < 40%. NCT06177041: IHC assay: unknown, CLDN18.2 cutoff: Moderate–High: 2/3+ ⩾40%, Low: 1/2/3+ ⩾10%+ and 2/3+ <40%. NCT05632939: IHC assay: DS-3, CLDN18.2 cutoff: Moderate–High: 2/3+ ⩾40%. NCT04805307: IHC assay: EPR19202, CLDN18.2 cutoff: High: 2/3+ ⩾20%. NCT05458219: IHC assay: 43-14A, CLDN18.2 cutoff: Moderate–High: 2/3+ ⩾20%. NCT03874897: IHC assay: 14F8, CLDN18.2 cutoff: High: 2/3+ ⩾40%, Low-Moderate: 2/3+ <40% or intensity 1+ with any percentage. NCT05458219: IHC assay: unknown.
Dose expansion cohort (n = 107).
ADC, antibody–drug conjugate; BsAb, bispecific antibody; CAR-T, chimeric antigen receptor-T; CRS, cytokine release syndrome; Gr, grade; H, CLDN18.2 expression high; ILD, interstitial lung disease; L, CLDN18.2 expression low; M, CLDN18.2 expression moderate; m, months; mAb, monoclonal antibody; mPFS, median progression-free survival; Nivo, nivolumab; ORR, overall response rate; PTX, paclitaxel; R, CLDN18.2 expression negative; RAM, ramucirumab; TRAEs, treatment-related adverse events.
FG-M108 is an afucosylated monoclonal antibody with enhanced ADCC activity. In a phase I/II trial, FG-M108 combined with CAPOX demonstrated an ORR of 81% in high CLDN18.2 expressors (2+ ⩾40%, n = 36) and 67% in low expressors (1+ ⩾10%, 2+ <40%, n = 15), with an mPFS of 11 months in high expressors. 64
ASKB589, targeting CLDN18.2-positive tumors (⩾1+), in combination with CAPOX and sintilimab, was reported with an ORR of 80% (n = 36). 65 A phase III trial in China is ongoing, including patients with CLDN18.2 expressing tumors (⩾2+, ⩾40% of tumor cells). In addition, over 10 monoclonal antibodies targeting CLDN18.2 are under clinical investigation.
Multimodal approaches targeting CLDN18.2
Various approaches to CLDN18.2-directed therapy are being developed.29,30
AZD0901, an antibody-drug conjugate (ADC) with an monomethyl auristatin E (MMAE) payload, demonstrated a confirmed ORR of 31% in a phase I trial involving patients with CLDN18.2 expression (⩾20% at 2+ intensity, n = 93), showing favorable results regardless of prior taxane or PD-1 therapy. At a 2.2 mg/kg dose, the treatment achieved the lowest GI toxicity and promising outcomes (ORR: 47%, mPFS: 4.8 months). 66 A phase III CLARITY-Gastric01 trial (NCT06346392) is currently underway to evaluate AZD0901 monotherapy versus physician’s choice chemotherapy in second-line or later settings. In addition, a phase II GEMINI-Gastric trial (NCT05702229) is evaluating its combination with bispecific antibodies targeting PD-1 × CTLA-4, PD-1 × TIGIT, and PD-1 × TIM-3 as the front-line setting. 67
IBI343, an ADC with topoisomerase I payload, utilizes Fc-silent antibodies to reduce ADCC and GI toxicity. A phase I trial (n = 159) reported primarily hematologic toxicities, with low rates of grade ⩾3 GI events (1.3%–1.9%). In patients with CLDN18.2 expression (with ⩾2+ intensity in ⩾40%), the ORR was 32.6%. Furthermore, the ORR was improved to 36.7% at 6 mg/kg and 47.1% at 8 mg/kg in patients with CLDN18.2-high expression (in ⩾75%). A phase III G-HOPE-001 trial (NCT06238843) is ongoing for patients with high-expression CLDN18.2 patients in later line. 68
Autologous CAR-T-cell product against CLDN18.2 (Satri-cel, CT041) has shown promising results in phase I study, including patients with CLDN18.2-expressing GC/GEJC (n = 51), achieving an ORR of 54.9%, a disease control rate (DCR) of 96.1%, and an mPFS of 5.8 months. Cytokine release syndrome (CRS) occurred in 96.9% of patients, but no grade ⩾3 cases were reported, and hematologic toxicities resolved within 14 days. 69 Recently, the randomized phase II trial of satri-cel (CT041) met its primary endpoint (PFS), which is higher than standard chemotherapy in China. 70
IBI389, a bispecific antibody targeting CLDN18.2 and CD3, enrolled 120 patients in its phase I trial. Initial efficacy data showed an ORR of 30.8%, a DCR of 73.1%, and an mPFS of 3.5 months in a subgroup of 26 patients with ⩾10% CLDN18.2 expression (IHC 2+/3+). Compared to other CLDN18.2-targeted therapies, GI toxicity rates were relatively low, and CRS occurred in 60% of patients, with grade ⩾3 CRS in less than 1%. 71 ASP2138 is another dual target agent with CLDN18.2 and CD3. Utilizing its advantage of reduced toxicity, ASP2138, in combination with standard chemotherapy, is under investigation for patients with CLDN18.2 expressing GC/GEJC or pancreas cancer (NCT05365581).
CLDN18.2 expression (IHC 2+/3+) is considered a predictive marker for patient selection in CLDN18.2-targeted therapy. Several attempts to expand the indication to a broader range of CLDN18.2-expressing gastric cancers are currently under investigation.
Dynamics of CLDN18.2 status
Examinations using surgical specimens have shown that CLDN18.2 expression tends to decrease from the superficial to the invasive front of primary tumors, 72 with intratumoral heterogeneity observed in approximately 38.5% of cases. 73 Furthermore, CLDN18.2 positivity rates tend to decrease in metastatic lesions. A Japanese study reported that the positivity rate was 28.6% in primary tumors compared to 20.2% in peritoneal metastases using a 75% cutoff criterion. The concordance rate of CLDN18.2 assessment between primary and metastatic sites was 75% (positive concordance rate 11.9%, negative concordance rate 63.9%). 72
In addition, a Korean study demonstrated varying CLDN18.2 positivity rates across different metastatic sites. Peritoneal lesions showed the highest positivity rate (44.3%) and positive concordance rate with the primary tumor (31.4%), while liver metastases exhibited the lowest positivity rate (17.9%) and positive concordance rate (12.8%). A paired analysis of primary and metastatic sites revealed discordant CLDN18.2 positivity results in 25.2% of patients. 73
Changes in CLDN18.2 expression before and after chemotherapy have been reported, with 40%–60% of cases showing a loss of expression when using a 75% cutoff criterion.42,45,47,72 By contrast, no data are currently available regarding changes in CLDN18.2 expression following zolbetuximab-containing treatment.
CLDN18 expression is regulated through transactivation by T/EBP/NKX2.1 (also known as TTF-1); however, methylation of the CLDN18 promoter region can inhibit TTF-1 binding, possibly leading to downregulation of CLDN18 expression.74,75 Chemotherapy-induced cytotoxic stress can generally promote DNA methylation, which may contribute to the decreased expression of CLDN18.2 observed during treatment.
Unlike HER2, whose downstream or compensatory pathways can sustain tumor regrowth, CLDN18.2 is not directly involved in proliferative signaling, so its loss may not directly lead to therapeutic resistance. Therefore, loss of CLDN18.2 expression may represent a transient phenomenon with different biological implications from the loss of HER2 due to clonal selection.
Trophoblast cell surface antigen 2
TROP2, encoded by the TACSTD2 gene, is a 36-kDa transmembrane glycoprotein also known as tumor-associated calcium signal transducer 2 or epithelial cell adhesion molecule 2. 76 This protein consists of a large extracellular domain, a single transmembrane domain, and a short intracellular tail. 77 The involvement of calcium transducers in cell proliferation, motility, and apoptosis suggests that TROP2 abnormalities may be associated with carcinogenesis. 78 It activates the ERK/MAPK cascade, 79 modulates the PI3K/AKT pathway, and interacts with β-catenin signaling80,81 (Figure 2). However, definitive evidence supporting its role as an oncogenic driver is lacking. The cancer-specific overexpression has made TROP2 an attractive target for cancer therapy. 77 Importantly, high TROP2 expression has been correlated with poor prognosis and aggressive disease in many cancer types, including GC/GEJC.82–84

TROP2 overexpression drives malignant cell proliferation and metastasis. TROP2 overexpression in malignant cells drives dysregulation of signaling pathways, including MAPK/ERK, PI3K/AKT, and β-catenin, leading to enhanced tumor proliferation, invasion, and metastasis. In addition, TROP2-mediated calcium signaling facilitates cell cycle progression, further contributing to tumor development.
TROP2 expression is observed in approximately 50% of GC/GEJC with moderate or strong intensity. 85 Several TROP2-directed therapies with ADCs have shown promising results in clinical trials for various solid tumors.86–88
Sacituzumab govitecan is an ADC targeting TROP2 by linking the antibody hRS7 to the topoisomerase I inhibitor SN-38 through a hydrolyzable linker called CL2A.89,90 Stress granule (SG) has been approved by the FDA for the treatment of metastatic triple-negative breast cancer, 86 hormone receptor-positive/HER2-negative metastatic breast cancer, 87 and metastatic urothelial carcinoma (UC). 88 An open-label, single-arm, multicenter phase Ib/II trial is ongoing to assess the efficacy of Sacituzumab govitecan for metastatic esophagogastric adenocarcinoma. 91
Sacituzumab tirumotecan (SKB264/MK-2870) is another TROP2-directed ADC developed using a belotecan-derivative topoisomerase I inhibitor as the payload, with a new linker that enables efficient payload release within tumor cells and the tumor microenvironment through pH-sensitive cleavage and enzymatic cleavage. In a phase II expansion cohort of the KL264-01 trial (NCT04152499), Sacituzumab tirumotecan (5 mg/kg, Q2W) showed an ORR of 22.0% and the DCR of 80.5% for previously treated patients with GC/GEJC. 92 The ORR was better in the second line (27.3%) than in the later line (15.8%). The median duration of response (DoR) was 7.5 months. Treatment-related adverse events (TRAEs) of Grade 3 or higher were reported in 52.1% of patients, which were mainly hematological toxicities. No cases of neuropathy or drug-related interstitial lung disease/pneumonitis were reported. To further evaluate the clinical potential of SKB264, several clinical trials are currently underway. Substudy 06C (NCT04644571) and 06D (NCT04644572), both phase I/II trials, are evaluating the safety and efficacy of SKB264 in advanced GC/GEJC, with 06C investigating its combination with pembrolizumab, and 06D comparing it to ramucirumab plus paclitaxel. In addition, the phase III trial TroFuse-015 (NCT06356311) is comparing SKB264 to physician’s choice treatments, including TAS-102, irinotecan, and paclitaxel. Furthermore, LIGHTBEAM-02A (NCT04826404), a phase I/II trial, is evaluating the safety and efficacy of SKB264 as monotherapy or in combination with chemotherapy for patients with colorectal cancer, pancreatic ductal adenocarcinoma, and biliary tract cancer.
Biomarker selection is essential for success in the large-scale randomized study. Membranous staining intensity by IHC did not serve as a biomarker in the previous study of sacituzumab tirumotecan. Similar ORR and DCR were reported in moderate to strong (25.0% and 75.0%) and weak to loss group (21.7% and 87.0%). 92 Datopotamab deruxtecan (Dato-DXd) is another TROP2-targeted ADC evaluated in the TROPION-lung 01 trial. 93 Notably, biomarker analysis of TROPION-lung 01 reported the utility of quantitative evaluation, including TROP2 expression in the cytoplasm using an artificial intelligence model (TROP2 QCS-NMR). This algorithm predicted the responders and non-responders of Dato-DXd. 94 The TROPION-PanTumor03 trial evaluates the safety and efficacy of Dato-DXd with or without chemotherapy for advanced solid tumors, including GC/GEJC (NCT05489211).
The underlying mechanisms of resistance are not yet fully understood; however, recent studies have suggested several contributing factors. 95 For example, loss of TROP2 expression may reduce the ability of ADCs to recognize cancer cells. Nevertheless, exploratory analysis from the TROPiCS-02 87 trial has indicated that the efficacy of sacituzumab govitecan is not dependent on TROP2 expression levels. In addition, mutations such as the TROP2 T256R variant, which impairs antibody binding, and the TOP1 E418K mutation, which affects the activity of SN-38, have been identified in breast cancer and may be involved in resistance. Further investigation is needed to clarify their relevance in GC/GEJC.
Nectin-4
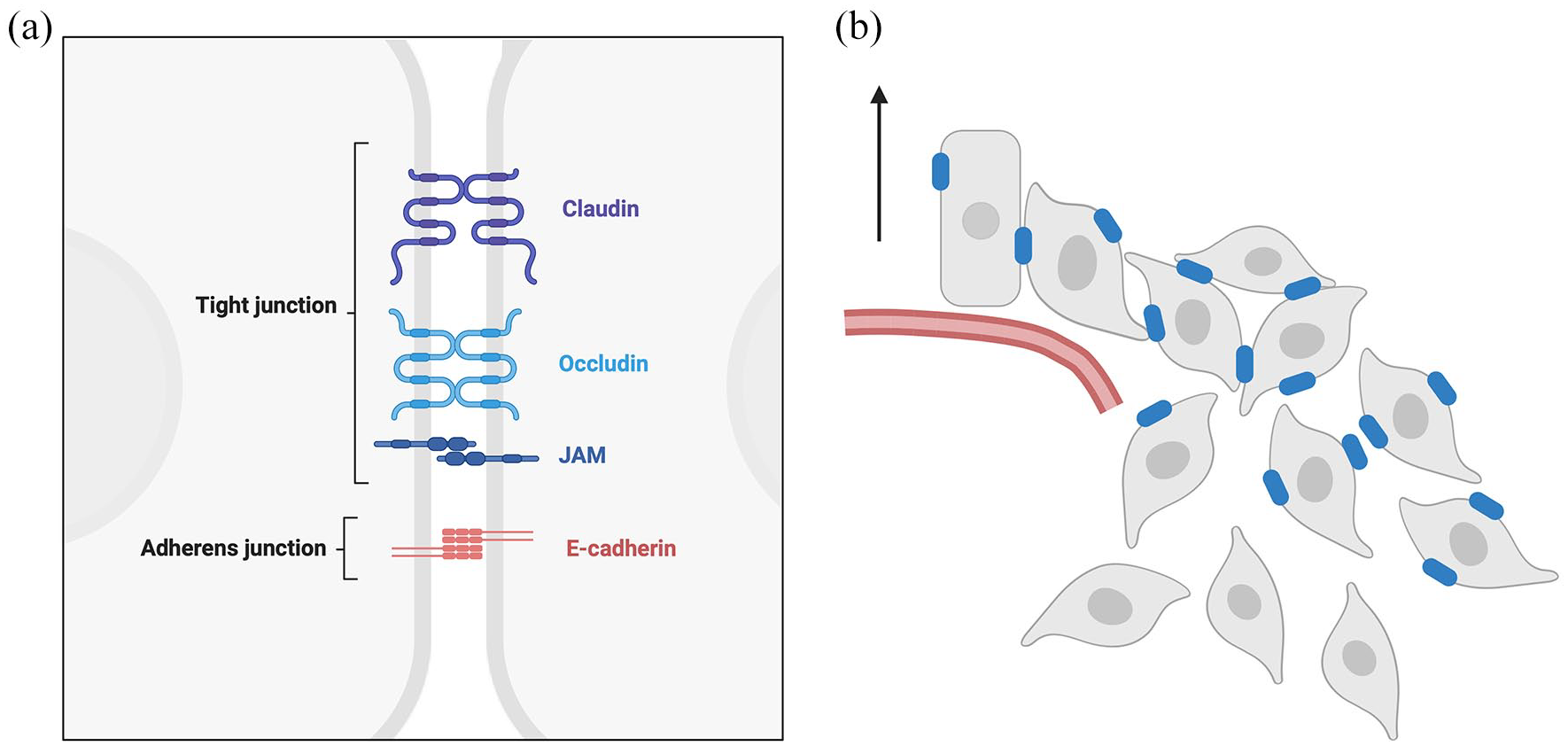
Nectin is an immunoglobulin-like molecule classified within the immunoglobulin superfamily, a subset of cell adhesion molecules that also includes cadherins, integrins, and selectins. Nectin is composed of four family members. Nectin possesses an N-terminal signal peptide, three immunoglobulin-like extracellular domains, a transmembrane region, and a cytoplasmic domain containing an afadin-binding motif. Unlike the others, Nectin-4 is predominantly expressed in embryonic and placental tissues, with its expression markedly decreasing in adulthood 96 (Figure 3). However, the overexpression of Nectin-4 in cancer has emerged as a promising focus for therapeutic targeting.97,98 Although both membranous and soluble Nectin-4 have been implicated in processes relevant to cancer, such as cell proliferation, invasion, angiogenesis, and immune evasion, its primary role is as a molecule associated with F-actin, interacting with afadin to contribute to cytoskeletal organization and cell adhesion.99–104 In addition, Nectin-4 binds to the T-cell immunoreceptor with Ig and ITIM domains (TIGIT), and TIGIT delivers inhibitory signals to T cells and NK cells. Dysregulation of this signaling pathway leads to decreased T cell activity, weakening the immune response against malignancies. 105
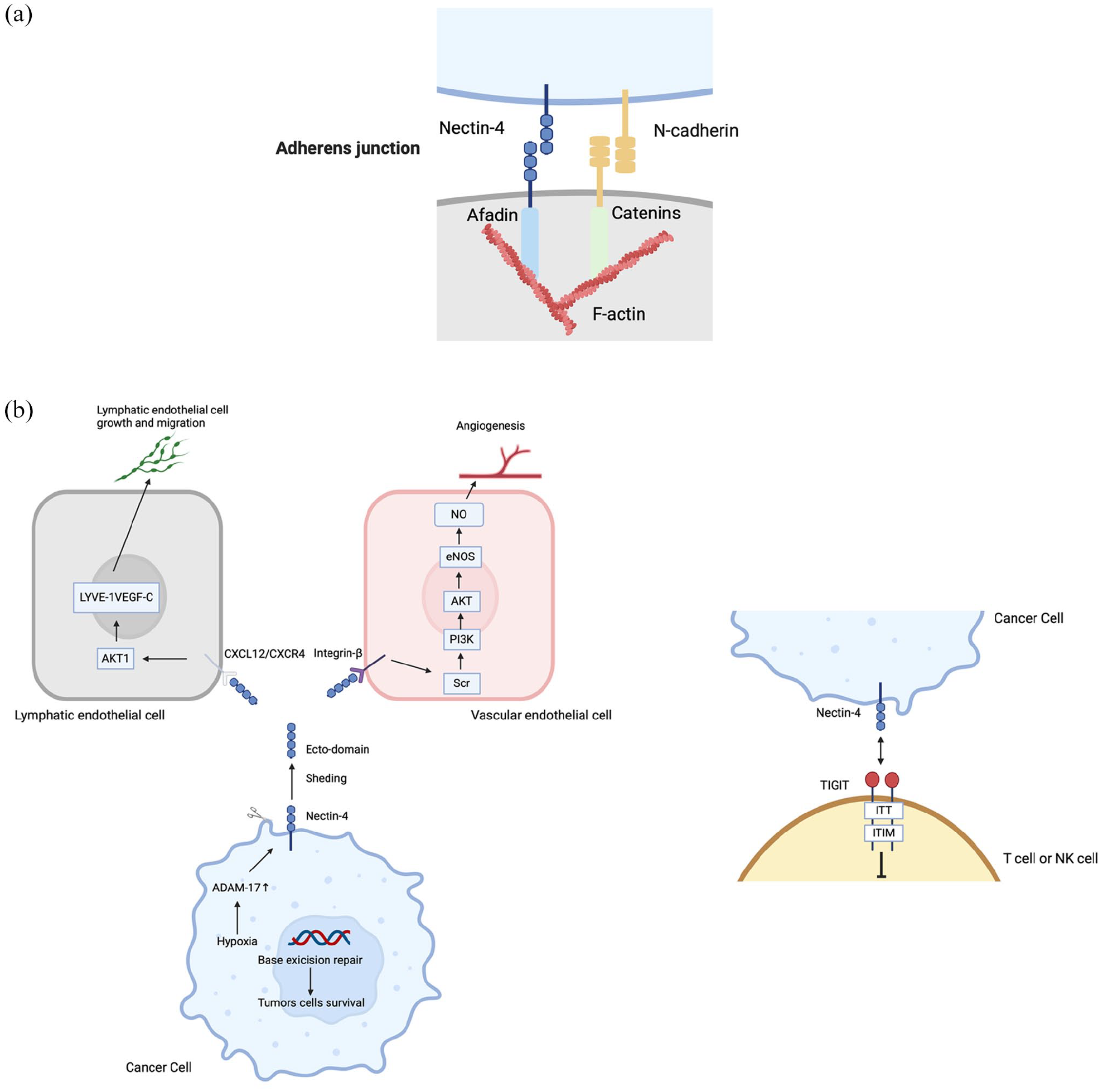
Nectin-4 signaling in normal and malignant cells. (a) In normal cells, Nectin-4 is localized at adherens junctions, where it interacts with afadin and F-actin to maintain cell–cell adhesion. This interaction is crucial for maintaining epithelial integrity. (b) In malignant cells, Nectin-4 undergoes ectodomain shedding, triggered by hypoxia-induced activation of ADAM-17. The released soluble Nectin-4 ectodomain interacts with CXCR4 on lymphatic endothelial cells or integrin β on vascular endothelial cells, promoting tumor progression. Specifically, CXCL12/CXCR4 signaling activates the AKT1 pathway in lymphatic endothelial cells, driving cell growth and migration. In vascular endothelial cells, Nectin-4 ectodomain binding initiates the Src/PI3K/AKT/eNOS pathway, enhancing NO production and angiogenesis. Furthermore, in cancer cells, hypoxia and the activation of ADAM-17 contribute to enhanced base excision repair and tumor cell survival, highlighting Nectin-4’s multifaceted role in oncogenesis and metastasis. Nectin-4, expressed in cancer cells, binds to TIGIT, which is primarily expressed in T cells and NK cells. Upon binding, this interaction triggers inhibitory signals through the ITT-like and ITIM (immunoreceptor tyrosine-based inhibitory motif) domains in the cytoplasmic region of TIGIT. These inhibitory signals suppress T cell and NK cell activation, thereby facilitating immune evasion by cancer cells.
While previous studies highlighted possible associations between Nectin-4 and cancer, similar to those observed for many other molecules, the considerable clinical outcomes of enfortumab vedotin (EV), a Nectin-4-targeting ADC in UC, have brought significant attention to its therapeutic potential. In the phase III trial EV-302/KEYNOTE-A39, the combination of EV and pembrolizumab significantly improved overall survival (31.5 months vs 16.1 months, HR 0.47 (95% CI 0.38–0.58), p < 0.001) and progression-free survival (12.5 months vs 6.3 months, HR 0.45 (95% CI 0.38–0.54), p < 0.001) compared to standard chemotherapy. 106 This trial established a new first-line therapy for platinum-sensitive cancers without requiring cisplatin.
Nectin-4 expression is moderate to high in GC and esophageal cancers. Nectin-4, as a cancer biomarker, has been elucidated to be intricately associated with lymph node metastasis mediated through the PI3K/AKT pathway, ultimately leading to an unfavorable prognosis, with its expression closely correlated with different TNM stages of GC.97,107
The EV-202 trial (NCT04225117) is a phase II study targeting adults with locally advanced or metastatic solid tumors. Patients received 1.25 mg/kg of EV on days 1, 8, and 15 of a 28-day cycle. As of March 3, 2023, the gastroesophageal adenocarcinoma (GEA) cohort included 42 patients, with 69% having received two or more prior systemic therapies. The confirmed ORR was 9.5%, which did not meet the threshold for promising activity. The DCR was 47.6%, the median DoR was 10.3 months, the PFS was 3.06 months, and the OS was 8.31 months. Common TRAEs included pruritus (n = 16, 38.1%), alopecia (n = 15, 35.7%), and dysgeusia (n = 13, 31.0%). Grade 3 or higher TRAEs included maculopapular rash (n = 3, 7%), neutropenia (n = 2, 5%), and hyperglycemia (n = 2, 5%). Notable TRAEs included skin reactions (n = 23, 55%), peripheral neuropathy (n = 7, 17%), and hyperglycemia (n = 3, 7%). EV demonstrated a safety profile. Thus far, no indications suggest that the remarkable efficacy observed in UC can be expected in GC/GEJC. Further investigation is warranted. 108
Although the mechanisms of resistance to EV remain unclear, multiple studies have demonstrated a decrease in membrane Nectin-4 expression in metastatic tissues, and reduced or absent membrane Nectin-4 expression in UC tissues may play a crucial role in EV resistance.109,110 However, some studies have confirmed the maintaining high expression of Nectin-4 in new tissue specimens after cancer recurrence. 111 This evidence highlights the complexity of EV resistance, suggesting that factors other than antigen reduction, such as intracellular signaling pathways or drug efflux mechanisms, may play a role. 112 The preclinical study reported that ABCB1, which encodes the ATP-binding cassette transporter multidrug resistance 1 (MDR-1)/P-glycoprotein (P-gp), was upregulated in EV-resistant tumors. Importantly, after P-gp was inhibited, sensitivity to EV was restored in in vitro and in vivo models. Furthermore, the combined use of a third-generation P-gp inhibitor and ADC was well tolerated in mice and successfully restored sensitivity to the ADC. Clinical data are required to clarify resistance mechanisms and develop effective strategies for patients undergoing EV therapy.
Caprin-1
Cytoplasmic activation/proliferation-associated protein 1 (Caprin-1) is an RNA-binding protein involved in various critical cellular processes. 113 Caprin-1 regulates cell cycle progression by modulating the expression of crucial genes such as CCND2 and c-myc, promoting the G1 to S phase transition and thereby enhancing tumor proliferation. 114 Cytoplasmic Caprin-1 migrates and accumulates in SGs in response to stressful stimuli, and cancer-specific membrane-expressed Caprin-1 is reported in various cancers, being identified as a novel and targetable molecule. 113 Under cellular stress conditions, Caprin-1 facilitates the formation of SGs via liquid–liquid phase separation (LLPS), helping cancer cells survive in harsh tumor microenvironments.115,116 These SGs sequester pro-apoptotic factors, contributing to protect cancer cells from stress-induced apoptosis. Caprin-1 regulates the Wnt/β-catenin pathway, promoting epithelial–mesenchymal transition (EMT) and maintaining cancer stem cell properties. 117 Membrane expression of Caprin-1 is observed in highly tumorigenic cancer stem cells and cells undergoing EMT (Figure 4). 118
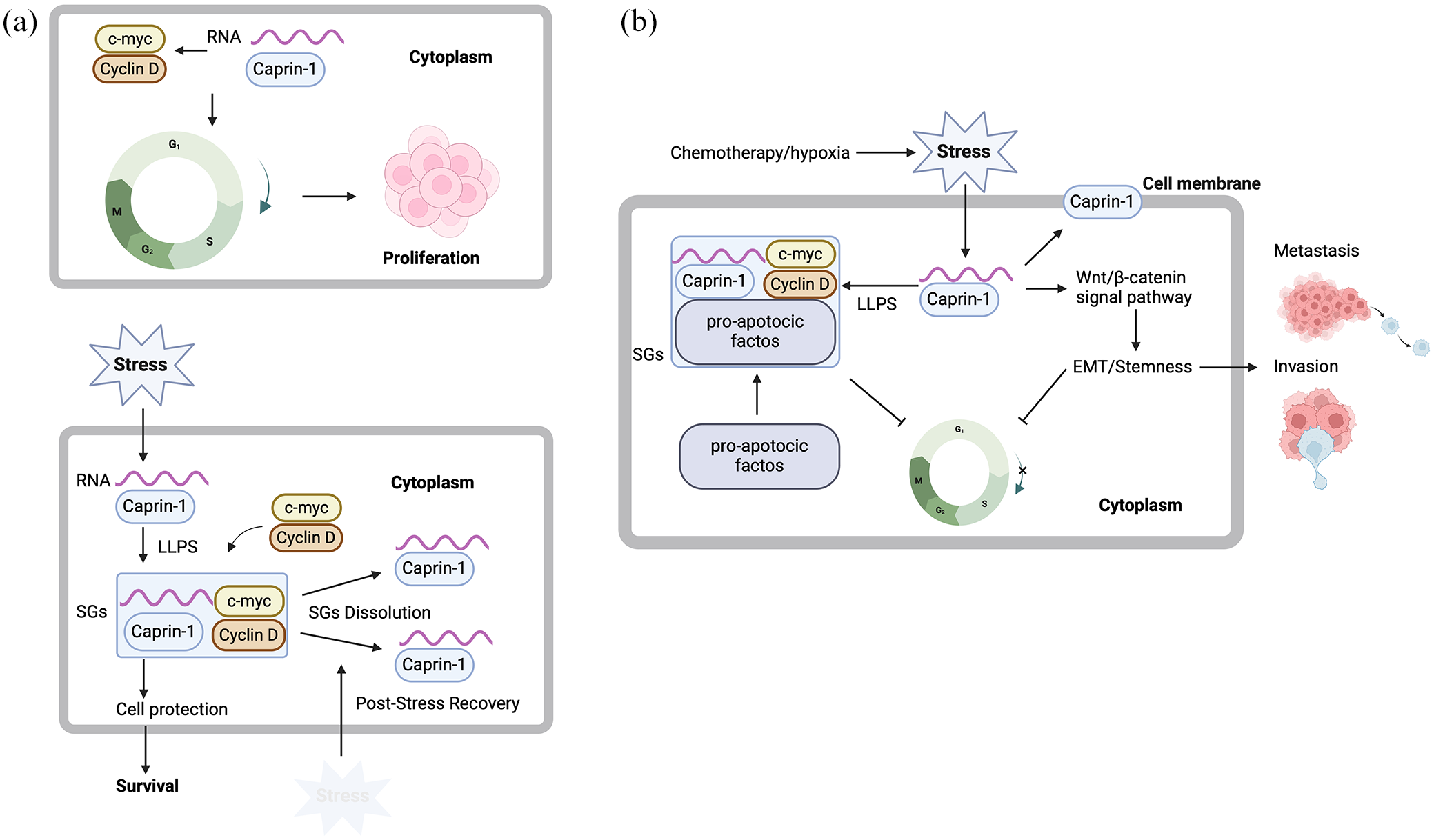
Caprin-1 functions in normal and malignant cells. (a) Caprin-1 regulates RNA stability and translation in normal cells, promoting cell cycle progression through the expression of Cyclin D and c-myc. This facilitates the transition from the G1 phase to the S phase, driving cell proliferation. Under stress conditions, cytoplasmic Caprin-1 undergoes LLPS to form SGs, protecting cells from stress and supporting survival. (b) In malignant cells, overexpression of Caprin-1 promotes tumor growth, survival, and metastasis. Cytoplasmic Caprin-1 accumulates in SGs and sequesters pro-apoptotic factors, protecting cancer cells from stress-induced apoptosis and contributing to chemotherapy resistance. Furthermore, Caprin-1 activates the Wnt/β-catenin signaling pathway, promoting EMT, stemness, and metastatic potential. Cancer-specific membrane-expressed Caprin-1 has been reported in various cancers, identifying it as a novel and targetable molecule.
TRK-950 is the first humanized IgG1 antibody targeting Caprin-1, which binds strongly and specifically to cancer cells and exerts antitumor effects by engaging immune cells like macrophages and NK cells.
In a phase I study (NCT02990481), no dose-limiting toxicities were observed at doses of 3–30 mg/kg IV weekly, and the maximum tolerated dose was not reached, confirming its safety and tolerability. A phase Ib study (NCT03872947) evaluated the combination of TRK-950 with standard-of-care regimens in previously treated patients with advanced cancers. In the GC/GEJC cohort (n = 9), the combination of TRK-950 on days 1 and 15 of a 28-day cycle) with ramucirumab and paclitaxel demonstrated the ORR of 55.6% and DCR of 100% without a new safety signal by adding TRK950. All four patients whose tumors had strong Caprin-1 expression (IHC 3+ in ⩾10% of tumor cells) achieved a partial response. 119 Currently, a two-stage design, open-label, randomized phase II trial is underway to evaluate the efficacy of TRK-950 (5 or 10 mg/kg) in combination with ramucirumab and paclitaxel as a second-line treatment for patients with Caprin-1 expressing GC/GEJC (IHC ⩾2+ in ⩾30% tumor cells).
Future directions
The identification of CLDN18.2 as a therapeutic target has expanded treatment strategies for GC/GEJC, accelerating the development of targeted therapies against tumor-associated molecules (Figure 5). Several agents targeting non-oncogenic drivers are currently under investigation, including tusamitamab ravtansine for CEACAM5, 120 SGN-B6A for integrin β6, 121 tisotumab vedotin for TF, 122 and mirvetuximab soravtansine for Frα. 123

Therapeutic strategies targeting non-oncogenic drivers in gastric cancer. Various therapeutic strategies targeting Claudin18.2, TROP2, NECTIN-4, and CAPRIN1 are currently under evaluation in clinical trials. Monoclonal antibodies, such as Zolbetuximab and TRK-950, induce tumor cell death through ADCC and CDC. Bispecific antibodies and T-cell engagers recruit immune cells, such as T cells, to tumor cells, enhancing immune-mediated cytotoxicity. ADCs, including Sacituzumab tirumotecan and Enfortumab vedotin, selectively deliver cytotoxic agents to tumor cells while minimizing the impact on normal tissues. CAR T cells, engineered to target Claudin18.2, utilize CD28 costimulatory and CD3ζ signaling domains to promote tumor cell death.
Recent advances in adoptive cell transfer using CAR technology offer a promising therapeutic platform for targeting tissue-associated molecules in GI cancers. 124 Guanylyl cyclase C (GUCY2C), a membrane-bound receptor that generates the second messenger cGMP, plays a crucial role in intestinal homeostasis and tumorigenesis. Building on the encouraging results of CLDN18.2-directed CAR-T therapy, GUCY2C-targeted CAR-T therapy has demonstrated promising efficacy in patients with GUCY2C-expressing colorectal cancer.125,126 In addition, CAR-T therapies targeting GPC3127–129 CEACAM5 130 and EpCAM 131 are under investigation for hepatocellular carcinoma and other GI malignancies.
Tumors with non-oncogenic driver alterations likely require multiple steps for carcinogenesis. CLDN18.2, TROP2, and Nectin-4 are involved in cell-to-cell adhesion, and their dysregulation may share common pathways associated with invasion, migration, and polarization. As targeted therapies for these biomarkers are developed, managing the overlap of multiple biomarkers will become a key challenge in optimizing treatment strategies.
Unlike genomic alterations such as receptor tyrosine kinase amplification or oncogene mutations, the regulation of non-oncogenic drivers is not genetically fixed. Instead, it is influenced by DNA methylation, transcriptional factors, and post-transcriptional modifications. The loss of expression is a potential key resistance mechanism to targeted therapies against non-oncogenic drivers. Qualitative transformations, such as EMT, may contribute to this downregulation; however, robust evidence linking EMT to resistance remains lacking. Addressing resistance mechanisms in targeted therapy for non-oncogenic drivers remains a significant challenge.
Conclusion
The emergence of CLDN18.2-directed therapy in GC/GEJC has underscored the significance of targeting non-oncogenic drivers, highlighting the need to move beyond genomic alterations alone. While these innovative strategies have demonstrated clinical efficacy, the therapeutic landscape is becoming increasingly complex. Key challenges include the development of precise predictive biomarkers, elucidating resistance mechanisms, and refinement of patient selection methods. A deeper understanding of genomic alterations and the tumor immune microenvironment will be essential for optimizing treatment outcomes. As novel therapeutic strategies continue to evolve, integrating these approaches with existing treatments will be critical for shaping the future of GC therapy. Addressing these challenges in an increasingly intricate treatment paradigm will be essential for advancing more effective and personalized therapeutic strategies.