
Abstract
Kirsten rat sarcoma (KRAS) mutations are present in up to 25% of non-small-cell lung cancer (NSCLC). KRAS G12C is the most common type of mutation, representing approximately half of the cases in KRAS-mutant NSCLC. Mutations in KRAS activate the RAF-MEK-ERK pathway, leading to increased cell proliferation and survival. Recent advances in drug development have led to the approval of KRAS G12C inhibitors sotorasib and adagrasib. This review explores the emerging therapeutic strategies in KRAS G12C-mutant NSCLC, including dual checkpoint blockade and combinations with checkpoint inhibitors, with a focus on the setting of advanced disease.
Plain language summary
In lung cancer, certain genes drive the growth of the disease. One common mutation is in the KRAS gene, specifically the KRAS G12C mutation, which occurs in about half of KRAS lung cancer. This mutation is particularly important because there are treatments that target this mutation. Immunotherapy, a treatment that boosts the immune system to help it recognize and attack cancer cells, has shown to be effective in this type of cancer. This review focuses on the latest treatment options for KRAS G12C, including immunotherapy and targeted KRAS G12C inhibitors.
Introduction
Kirsten rat sarcoma (KRAS) oncogene is the most common oncogene driver mutation in cancer and is found in approximately 25% of non-small-cell lung cancers (NSCLC). In the year 1982, it was the first human oncogene to be discovered. 1 Despite its early identification, KRAS is known as the “undruggable target” due to the lack of classic drug target sites, its high affinity for GTP, and its unusual shape. Recent progress in drug development led to the design of small-molecule inhibitors that target KRAS G12C mutations. This game-changer has opened the door for new treatment options with recent approvals of AMG510 (sotorasib) and MRTX849 (adagrasib).2,3 This review will focus on KRAS G12C-mutated NSCLC, exploring its role in tumor growth as an oncogene driver mutation, as well as current and future therapeutic strategies that involve immunotherapy.
KRAS is a RAS protein that is part of the small GTPase family. 4 These proteins are intracellular guanine nucleotide-binding proteins (G proteins) that regulate cell growth and survival. 4 It shifts cyclically between an active GTP-bound state and an inactive GDP-bound state. Two important regulatory proteins control this shifting process: guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). 5 When the GEFs interact with the KRAS-GDP complex, the KRAS-GDP complex has decreased affinity to GDP and GDP is replaced with GTP. GTP has higher affinity and concentration than GDP. The KRAS-GTP complex acquires a change in conformation in switches I and II of the G domain leading to KRAS activation. 6 On the other hand, GAPs promote binding between KRAS and GDP by promoting the GTPase activity of KRAS to maintain the inactive GDP-bound state.
When KRAS is activated, it triggers multiple downstream signaling pathways that drive cancer growth. The RAF-MEK-ERK pathway is the main downstream pathway of KRAS signaling. Activated KRAS-GTP leads to phosphorylation events involving MEK1/2 and ERK1/2. These events regulate the transcription and translation of specific genes, subsequently influencing cell proliferation and survival. 7
Another pathway involved is the PI3K-AKT-mTOR pathway which is crucial for cellular growth, metabolism with glucose transportation, and apoptosis. 8 KRAS can activate PI3K which leads to downstream activation of AKT and mTOR proteins. Activation of mTOR proteins leads to cell proliferation and survival. AKT phosphorylates and activates Bcl-XL/Bcl-2-associated death promoters, eventually inhibiting apoptosis. 9
RAL guanine nucleotide dissociation stimulator is another downstream signaling KRAS protein. It functions as a GTP/GDP exchange factor to promote conversion from RAL GDP to RAL GTP.10,11 The downstream factors from RAL proteins include Rac/cell division cycle 42 (CDC42) for cell migration, TANK-binding kinase 1 associated with viral immunity, and phospholipase D involved with endocytosis10,11 (Figure 1).
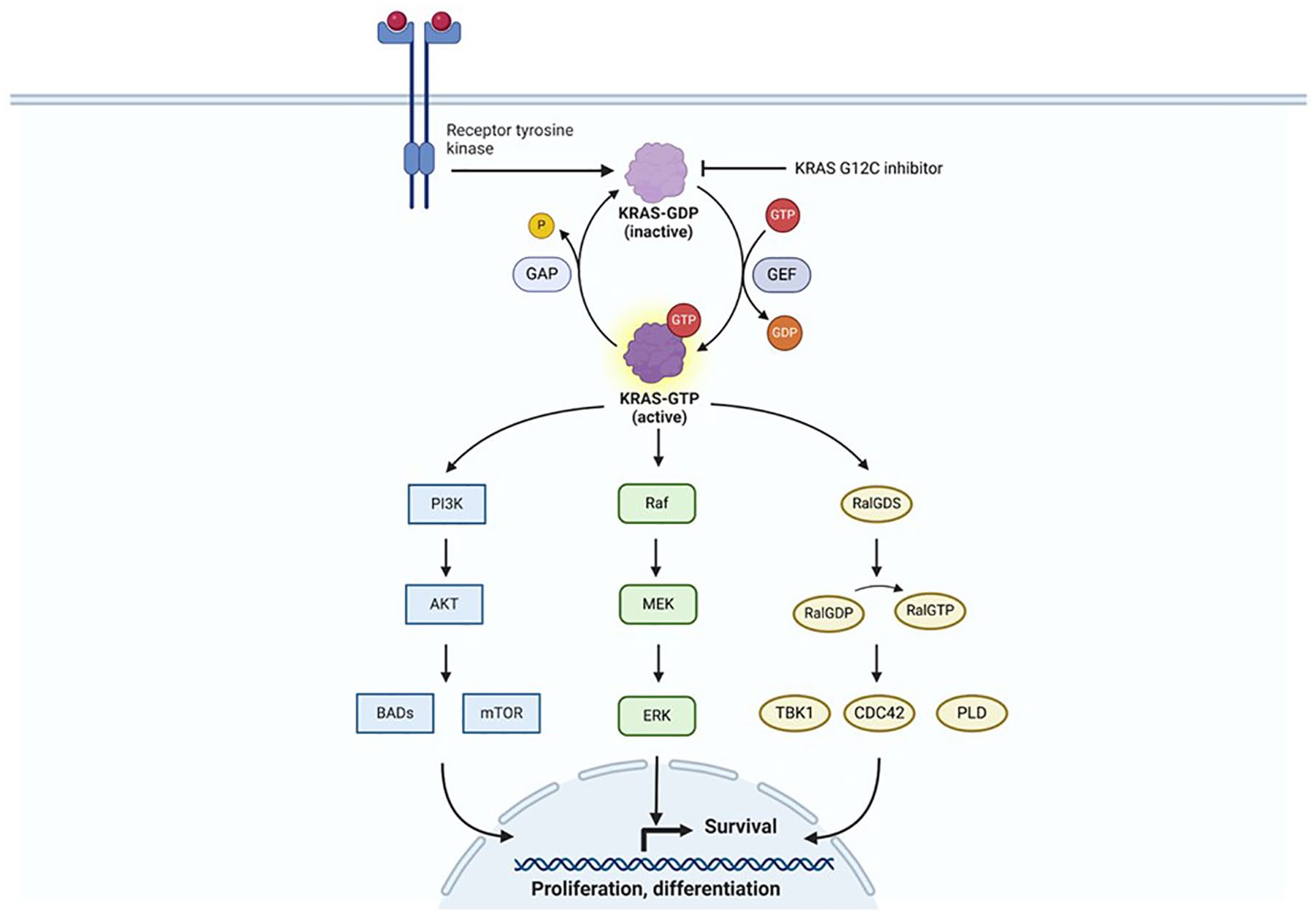
Schematic illustration of the KRAS signaling pathway. KRAS activation leads to downstream signaling through the RAF-MEK-ERK, PI3K-AKT-mTOR, and RalGDS pathways.
Mutations in KRAS, particularly at codons 12 (G12), 13 (G13), and 61 (Q61), affect the protein’s GTPase activity. As mentioned above, these mutations lead to continuous activation of the signaling pathways. 12 KRAS mutations are common in NSCLC, pancreatic adenocarcinoma, and colorectal cancer. The KRAS G12C mutation is the most common type of KRAS-mutated lung cancer. 13 Up to 80% of patients with KRAS-mutant NSCLC have a smoking history, in comparison to KRAS G12D, which is more frequent in non-smokers. KRAS G12C is more common in women who are often younger with less smoking history than men. 14
Rationale for immunotherapy in KRAS-mutated NSCLC
The use of immunotherapy with immune checkpoint inhibitors such as pembrolizumab, nivolumab, cemiplimab, and atezolizumab has transformed the treatment landscape in lung cancer, improving survival in patients with advanced and now early-stage disease.15–21
The presence of a KRAS mutation is associated with an inflamed tumor microenvironment (TME) and increased tumor immunogenicity. 22 KRAS-mutant tumors have greater T-cell infiltration compared to KRAS-wild-type tumors. 22 This contributes to enhanced response to immunotherapy. KRAS-mutant tumors have an increased proportion of programmed death-ligand 1+ (PD-L1+)/TIL+ suggesting an inflammatory phenotype and adaptive immune resistance. 22 A study has shown KRAS mutation leads to upregulation of PD-L1 through the p-ERK signaling pathway. 23 Mutation profile analysis showed that KRAS mutations are associated with increased tumor mutational burden (TMB) and immunogenicity. 22 High TMB reflects genomic instability leading to increased neoantigen production that can attract immune cells. 24 In addition, high TMB has been linked to smoking and alteration in DNA replication and damage repair genes in KRAS-mutant NSCLC.24–26
KRAS mutations are typically mutually exclusive of other actionable alterations such as EGFR and ALK. There is heterogeneity in clinical outcomes of patients with KRAS-mutant NSCLC based on co-occurring alterations. The most common co-mutations in KRAS-mutant NSCLC are STK11 (serine/threonine kinase 11), KEAP1 (Kelch-like ECH-associated), TP53, SMARCA4, and CDKN2A/CDKN2B. 27 Each of these co-mutations can influence the biology and immune microenvironment of the cancer (Figure 2).

Summary of the effects of co-mutations on the tumor microenvironment in KRAS-mutant NSCLC.
STK11, also known as LKB1, acts as a tumor suppressor gene that regulates cell metabolism and growth by phosphorylation of adenosine monophosphate-activated protein kinase (AMPK) and 12 AMPK-related kinases.28,29 Loss of STK11 in KRAS-mutant tumors promotes interleukin-6 (IL-6) production which attracts neutrophils, decreases T-cell infiltration, and leads to higher T-cell exhaustion. 30 NSCLCs with STK11-loss also express low levels of PD-L1. 31 The inactivation of STK11 leads to an “immune-cold” TME and is associated with poor response to immunotherapy.28,30,32 STK11 co-mutation occurs in approximately 10%–28% of KRAS G12C-mutant NSCLC. 33
KEAP1 protein is a negative regulator of the NRF2 pathway, which is involved in the oxidative stress response by triggering antioxidant and anti-inflammatory effects. 34 Mutations in KEAP1 lead to NRF2 activation, promoting cell growth and chemoresistance. KEAP1 mutations occur in about 11% of NSCLC. 35 KRAS-mutant NSCLC with concurrent KEAP1 and/or STK11 mutations had worse outcomes compared to those without KEAP1 and STK11 mutations. The presence of KEAP1 mutations alone or in combination with STK11 mutations is associated with worse progression-free survival (PFS) and overall survival (OS), even with the use of chemo-immunotherapy. A study showed that the median OS for patients with KRAS/STK11 double-mutant NSCLC was 12 months compared to KRAS-mutant only NSCLC was 21 months (hazard ratio (HR) 1.7, 95% confidence interval (CI): 1.1–2.4, p = 0.002). 27 In the same study, KEAP1 co-mutation had a median OS of 10 months (HR 2.1, 95% CI: 1.4–3.1, p < 0.0001). 27
KRAS mutations influence the TME, leading to immune evasion and reduced effectiveness of therapies especially immunotherapy.36,37 KRAS mutations promote immune escape by producing pro-inflammatory cytokines and chemokines, such as IL-6, IL-8, and GM-CSF. 38 These factors attract immunosuppressive cells like myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) into the TME. This inhibits the immune response against the tumor. KRAS-mutant tumors have increased expression of PD-L1, which interacts with the PD-1 receptor on T cells to suppress their antitumor activity. 39 These tumors typically have reduced infiltration of cytotoxic T lymphocytes (CTLs) essential for effective antitumor response and have increased infiltration by immunosuppressive cells such as MDSCs and Tregs.37,40
KEAP1 and STK11 mutations are associated with an adverse immunosuppressive microenvironment, creating resistance to immune checkpoint inhibitors. This microenvironment is characterized by the depletion of CD8+ cytotoxic T cells while preserving the CD4+ effector subsets. 41 Adding CTLA4 blockade to PD-(L)1 inhibition might mitigate this resistance. The combination of PD-(L)1 and CTLA4 inhibition utilizes two critical aspects of the suppressive STK11MUT and/or KEAP1MUT NSCLC tumor immune microenvironment. The first is the retention of anti-CTLA4-responsive CD4+ T cells, including TH1 T cells. The second is the reprogramming of myeloid-cell-rich tumor ecosystem to inducible nitric oxide synthase (iNOS)-expressing tumors in response to dual immune checkpoint blockade (ICB). 41
Emerging evidence suggests that STK11 and KEAP1 mutations are not equivalent in effect and that predicting immunotherapy outcomes solely based on the presence or absence of these mutations may be insufficient. 42 For example, truncating mutations in STK11 are more frequent in exons 1 and 2 which are associated with a worse prognosis compared to mutations in exons 3–9. 43 KEAP1 mutations are distributed throughout the gene and less clustered in specific hotspot regions. 35 The functional impact of these missense mutations remains unknown and suggests the need for more accurate classification based on pathogenicity, protein expression, and downstream effector activity. 42 Mutation clonality can also impact immunotherapy efficacy. Clonal mutations with loss of heterozygosity in KEAP1, unlike subclonal mutations, are associated with resistance to PD-(L)1 monotherapy and a T-cell-excluded microenvironment. 44 The observation that some STK11 and KEAP1-mutated tumors may still respond to ICI highlights the importance of identifying factors that can guide clinical decisions. For example, for those without concurrent KRAS mutations with subclonal STK11 or KEAP1 mutations and high PD-L1 expression levels, PD-(L)1 monotherapy may be sufficient. Alternatively, those with low or negative PD-L1 expression, concurrent KRAS mutation, truncating mutations, or clonal mutation may require PD-(L)1 inhibitor combination therapies with chemotherapy or CTLA4 inhibitor. 42
A SMARCA4 co-mutation occurs in nearly 6% of KRAS-mutant lung cancers. 45 SMARCA4 encodes the SWItch/Sucrose NonFermentable complex, which controls gene expression by altering chromatin structure.46,47 The loss or mutation of SMARCA4 disrupts chromatin remodeling and may dysregulate transcription programming. 48 Lung adenocarcinomas with KRAS-SMARCA4 co-mutations, treated with or without immunotherapy, have poor survival outcomes.45,49 These co-mutations were found to have lower proportions of CD8 and activated CD4+ T cells than those found in KRAS-mutant only and KRAS-TP53-mutant lung cancers, indicating an immunosuppressive TME. 45
Tumors with both KRAS and TP53 co-mutations in NSCLC have demonstrated high TMB, high PD-L1, and high tumor cell proliferation. 50 Genome-wide expression analysis showed that KRASmut/TP53mut overexpress the CX3CL1 gene. In its soluble form, the CX3CL1 chemokine (additionally termed fractalkine) can attract immune effector cells such as tumor-infiltrating CD8+ T cells, natural killer cells, and dendritic cells to the tumor leading to antitumor immune effect.51,52 In lung adenocarcinoma, increased mRNA expression of CX3CL1 was associated with improved OS. 52 This gene is also associated with increased myeloid diversity and improved response to immunotherapy; thus, it could potentially be a biomarker of response in lung cancer. 53
Testing for KRAS mutations by genomic profiling is recommended in NSCLC. 54 Liquid biopsy for plasma NGS testing can provide a noninvasive option for detecting these mutations. Routine testing for co-alterations such as STK11, KEAP1, and TP53 mutations is not yet recommended by clinical guidelines, though many NGS vendors already include this information in their NGS profile reports. While not yet standard in clinical practice, these mutations are increasingly being recognized as prognostic biomarkers and may eventually guide treatment decisions.
KRAS inhibition
AMG-510 and MRTX849 are the two pioneers of allosteric inhibitors for KRAS G12C. These inhibitors can form covalent bonds with the mutant cysteine in codon 12 to inhibit GDP-GTP exchange and maintain the KRAS protein in its inactive state, preventing downstream signaling pathways.55,56 The drugs not only have direct anticancer activity but they also exhibit immune-modulatory effects that contribute to their efficacy. Preclinical studies of AMG-510 demonstrated that this drug increases the infiltration of T cells, mainly CD8+ T cells, into the tumor. It induces a pro-inflammatory microenvironment marked by increased interferon signaling, chemokine production, antigen processing, cytotoxic and natural killer cell activity, along with markers of innate immune system activation. The inflamed TME renders the tumor more responsive to immune checkpoint inhibition. 57
Resistance to KRAS inhibitors represents a challenge in the treatment of KRAS-mutant NSCLC. Many patients demonstrate primary resistance and never respond to the molecules, while other patients may initially respond but subsequently develop resistance leading to treatment failure and disease progression. The mechanisms of resistance to KRAS inhibition are multifaceted. Studies have shown that co-occurring mutations in KEAP1, SMARCA4, and CDKN2A are associated with poor OS in those with NSCLC treated with sotorasib or adagrasib.58,59 Regarding secondary resistance, in one study by Awad et al., 60 genomic and histologic analyses were performed using tumor biopsy samples of those who developed resistance after treatment with adagrasib monotherapy in KRAS G12C-mutant cancer. In 45% (17/38) of patients, a possible mechanism of resistance was identified such as a secondary KRAS mutation including KRASR68S, KRASH95D/G/N/R, and KRASY96C/D/H/N or amplifications in KRAS. Alternative oncogenic alterations that activate the RTK-RAS signaling pathway, without affecting KRAS, were seen, as were two cases of histologic transformation from lung adenocarcinoma to squamous-cell carcinoma. However, in the majority of patients, no new genomic event was detected to explain the pattern of resistance. 60
Current strategies: Immunotherapy for KRAS G12C NSCLC
As of the writing of this article, patients with KRAS G12C mutations are treated in the frontline with non-selected, immunotherapy-based treatments, with or without chemotherapy. 61 KRAS G12C inhibitors are currently used in the second line as monotherapy after both demonstrated an improvement in PFS over docetaxel in randomized clinical trials.62,63 Many retrospective studies have examined the efficacy of currently available immunotherapy-based regimens specifically in the KRAS-mutated patient population.
Single-agent immunotherapy
For patients with metastatic NSCLC with PD-L1 ⩾50%, regardless of squamous or non-squamous histology, there are currently three available single-agent immunotherapies that are approved by the European Medicines Agency (EMA) and the Food and Drug Administration (FDA): pembrolizumab, atezolizumab, and cemiplimab.15,16,18,64–66
Regarding the efficacy of immunotherapy specifically in the KRAS-mutated NSCLC, a subgroup analysis of patients enrolled on KEYNOTE-042 has been performed. 67 KEYNOTE-042 randomized patients with PD-L1 expression ⩾1% to either single-agent pembrolizumab or chemotherapy. 68 A subgroup analysis of outcomes by KRAS mutational status included only 12 patients with a KRAS G12C mutation. 67 While this group was small, the ORR was 66.7% (95% CI: 34.9–90.1), mPFS 15 months (95% CI: 10–NR), and mOS not reached (95% CI: 23–NR), suggesting these patients did not perform worse than those without KRAS G12C mutations. 67 Additional retrospective data have suggested that PD-L1 expression, both >1% and ⩾50%, is particularly predictive for ICI efficacy in patients with KRAS mutations. 69
Several groups have published conflicting results regarding the efficacy of immunotherapy for KRAS G12C versus KRAS non-G12C. One single-center retrospective analysis performed at Memorial Sloan Kettering indicated that patients treated with first-line immunotherapy may have worse outcomes for those with KRAS G12C when compared with those with KRAS non-G12C mutations. 33 Landmark 12-month PFS was 59% (22/37) in patients with non-G12C subtypes, compared with only 29% (11/38) with G12C mutations. 33 This analysis was not designed to compare outcomes with patients without KRAS mutations.
However, additional data have suggested that for patients with a PD-L1 expression ⩾50%, KRAS G12C mutations were associated with improved outcomes with first-line single-agent immunotherapy when compared to either patient with KRAS non-G12C mutations or KRAS wild-type tumors.70–72 Among 696 patients from the German National Network Genomic Medicine Lung Cancer who were PD-L1 ⩾50%, patients with a KRAS G12C mutation demonstrated a longer PFS (25.3 (14.3–36.2) months) compared with non-G12Cmut KRAS (9.6 (7.1–12.1)) and KRASwt patients (10.3 (8.0 = 12.5) months). For patients with a KRAS G12C mutation, a TP53 co-mutation predicted an increased response rate (69.7% vs 46.5%) for TP53-mutated (TP53mut) versus wild-type as well as PFS (HR = 0.59, p = 0.009), with a trend toward improvement in OS (HR = 0.72, p = 0.16). 19 For patients with KRAS G12C/TP53 co-mutations with a PD-L1 ⩾50%, there was an impressive mPFS and mOS (33.7 and 65.3 months, respectively). 19 An additional data set demonstrated that TP53/KRAS co-altered NSCLC derived significant clinical benefit from PD-1 inhibitors, more than either the KRAS-mutated/TP53 wild-type or KRAS wild-type groups. However, this analysis was not restricted to KRAS G12C specifically. 73 A similar multicenter study from France of 681 non-squamous patients with PD-L1 ⩾50%, which included 86 patients with KRAS G12C mutations, demonstrated a mPFS of 7.0 months (3.7–14) for KRAS G12C with a mOS of 18.4 (12.6–NR) months, with similar results seen in both KRAS non-G12C and wild-type KRAS. 74
We anticipate future data will more accurately describe efficacy outcomes in this patient population, given there are a number of prospectively enrolling clinical trials detailed below of frontline treatment of patients with KRAS G12C with single-agent immunotherapy control arms. In the interim, however, these data cumulatively show that frontline immunotherapy is efficacious for patients with KRAS G12C mutations and that alterations such as TP53 can potentially predict patient outcomes.
Single-agent immunotherapy in combination with chemotherapy
For patients with PD-L1 0%–49%, there are multiple combinations of chemotherapy plus single-agent immunotherapy that have demonstrated significant clinical benefit and are FDA- and EMA-approved. Pembrolizumab plus chemotherapy, cemiplimab plus chemotherapy, and atezolizumab plus chemotherapy with or without bevacizumab all have demonstrated improvements in patient outcomes over chemotherapy alone.75–79 The majority of patients with KRAS G12C mutations with PD-L1 0%–49% will be treated with one of these regimens, similar to those patients without KRAS G12C mutations. Understanding how patients with KRAS G12C mutations specifically perform using these regimens has never been prospectively studied. However, similar to single-agent immunotherapy, multiple retrospective analyses have been conducted that were either subgroup analyses of clinical trials or multicenter retrospective cohort studies to better inform both our prognostic and predictive understanding of these patients.
A subgroup analysis of KEYNOTE-189 of patients treated with pembrolizumab plus chemotherapy included 26 patients with a KRAS G12C mutation, who demonstrated an ORR of 50% versus 47.5% in patients without any KRAS mutation. 80 Progression-free survival was 11 months (95% CI: 6–18), while it was 9 months (95% CI: 7–14) for those without KRAS mutations. 80 Median OS was 18 months (95% CI: 11–NR) with KRAS G12C, while it was 23 months in those without KRAS mutations. 80
In a multicenter study of 138 patients with KRAS G12C treated with first-line chemo-immunotherapy, ORR was 41% (95% CI: 32–41), mPFS was 6.8 months (95% CI: 5.5–10), and mOS was 15 months (95% CI: 11–28). 81 When compared with patients with non-G12C KRAS mutations, in an adjusted analysis, there was not a statistically significant difference in PFS (HR 0.82, 95% CI: 0.61–1.11, p = 0.2) or OS (HR 0.55, 95% CI: 0.30–1.01, p = 0.053). 81 These data demonstrate that patients with KRAS G12C mutations can derive significant benefits from treatment with combination chemo-immunotherapy. We will obtain prospective data on the effectiveness of chemoimmunotherapy for patients with KRAS G12C mutations from the control arms of the upcoming phase III, randomized, first-line trials investing novel agents in the previously untreated setting, such as the CodeBreak202 trial. 82
Doublet immunotherapy with or without chemotherapy
Doublet immunotherapy combinations currently available include nivolumab plus ipilimumab with or without chemotherapy, as well as tremelimumab plus durvalumab with chemotherapy.83,84 Subgroup efficacy analysis has been performed for patients with KRAS mutations for CheckMate 227, CheckMate 9LA, and POSEIDON, though none of these analyses differentiate between KRAS G12C and non-G12C.83,85–87
The CheckMate-227 trial compared ipilimumab plus nivolumab to chemotherapy. 84 In the KRAS mutant population, ipilimumab/nivolumab outperformed chemotherapy with a mOS of 17.5 versus 15.7 months (HR 0.79, 95% CI: 0.55–1.12). 85 With the addition of two cycles of chemotherapy in CheckMate 9LA, at the 3-year update, patients with a KRAS mutation demonstrated an improvement in mOS at 19.2 versus 13.5 months (HR 0.72, 95% CI: 0.48–1.08). 87
POSEIDON was a three-arm randomized trial of tremelimumab plus durvalumab plus chemotherapy, durvalumab plus chemotherapy, or platinum chemotherapy alone. In the KRAS-mutated subgroup, tremelimumab, durvalumab, and chemotherapy demonstrated an impressive mOS of 25.7 months compared with 10.4 months with chemotherapy (HR 0.56, 95% CI: 0.36–0.88). 86 Similarly in the durvalumab, tremelimumab, and chemotherapy arm, mPFS was 8.5 versus 4.7 months with chemotherapy alone (HR 0.57, 95% CI: 0.35–0.92). 86 Based on the results of these three trials of dual checkpoint blockade, the combination of doublet immunotherapy plus chemotherapy could be considered for patients with KRAS G12C mutations, though with the disclaimer that none of these data sets differentiated between KRAS G12C and KRAS non-G12C.
One of the biggest limitations of using dual checkpoint blockade is the increased risk of immune-related adverse events (irAEs) over single-agent immunotherapy regimens. However, as the science of predicting which patients are at higher risk for adverse events evolves, dual checkpoint blockade may be able to be used with decreased toxicity. As an example, germline single-nucleotide polymorphism data were used in a cohort of patients to develop and validate polygenic risk scores for hypothyroidism, which could predict the development of thyroid irAEs (HR per SD = 1.34; 95% CI: 1.08–1.66; AUROC = 0.6). 88 In a separate study, distinct human lymphocyte antigen-DR alleles were associated with specific immunotherapy-related adverse events, such as type 1 diabetes or hypophysitis. 89 Utilizing genetic biomarkers to better understand toxicity risks will allow for informed patient discussions and shared decision-making regarding the optimal treatment strategy.
Effect of co-mutations on immunotherapy efficacy
STK11 or KEAP1, occurring either independently or as co-mutations concurrent with KRAS, is increasingly being recognized as both prognostic and perhaps also predictive. 90 STK11 co-mutations occur in 28% of patients with KRAS G12C mutations, and KEAP1 occurs in 23%. 33 Co-mutations in STK11 or KEAP1 have both been demonstrated to be associated with inferior outcomes with immunotherapy in patients with KRAS G12C.27,32,44,58
As a specific example, in a combined cohort from four medical centers of 1261 patients, STK11 was associated with significantly worse PFS (HR 2.04, p < 0.0001) and OS (HR 2.09, p < 0.0001). 31 Similarly, KEAP1 demonstrated inferior outcomes both of PFS (HR = 2.05, p < 0.0001) and OS (HR 2.24, p < 0.0001). 31 Interestingly, the inferior outcomes of STK11 and KEAP1 were observed only in patients with co-mutations in KRAS, but not in patients who had KRAS wildtype. 31 Similar results were found also for patients treated with chemotherapy alone. 91
In KEYNOTE-189, whole-exome sequencing data from both tumor and normal DNA were evaluable for 289 (47%) of 616 patients, of whom 54 (19%) had an STK11 mutation and 68 (24%) had a KEAP1 mutation; 29 (10%) had both STK11 and KEAP1 mutations. PD-L1 Tumor Proportion Score (TPS) tended to be lower in patients with versus without STK11 mutation (median (IQR) 0% (0–16) vs 15% (0–75)), whereas TMB score tended to be higher in patients with mutation (209 (132–265) vs 146 (89–264)). Similar patterns were seen for patients with or without KEAP1 mutation (PD-L1 TPS: 1% (0–13) vs 20% (0–75); TMB: 173 (124–267) vs 147 (89–263)). Although the ORR of pembrolizumab plus chemotherapy was lower and PFS and OS shorter in patients with versus without STK11 and KEAP1 mutation, pembrolizumab plus chemotherapy was associated with numerically better outcomes than placebo plus chemotherapy regardless of mutation status, and the 95% CIs were wide given the modest mutation frequency and the 2:1 randomization in favor of pembrolizumab plus chemotherapy for patients with metastatic nonsquamous NSCLC, regardless of STK11 or KEAP1 status. 92
A recent publication from Skoulidis et al. 41 demonstrated that for patients with STK11 and/or KEAP1, the addition of a PD-L1 inhibitor durvalumab did not improve outcomes over chemotherapy; however, dual checkpoint blockade with the addition of tremelimumab, a CTLA4 inhibitor, to the PD-L1 and chemotherapy combination showed clinical benefit.
This strategy will be studied prospectively with the TRITON study, which is enrolling patients with metastatic non-squamous NSCLC with KRAS mutations and/or STK11 and/or KEAP1. 93 Patients will be randomized to either tremelimumab, durvalumab, and chemotherapy versus pembrolizumab plus chemotherapy. 93
Moving forward, as the next wave of clinical trials is designed for patients with KRAS mutations, consideration should be given to the stratification of patients by specific allele or co-mutation status. Stratifying based on STK11 or KEAP1 co-mutations may allow for a more nuanced understanding of treatment efficacy, as well as a more personalized treatment approach. Prospectively designing trials that are statistically powered to understand the effect of these co-mutations will allow for a greater understanding of small, unplanned, retrospective analyses, to guide future clinical decision-making and design more effective treatment paradigms.
Combining KRAS G12C inhibition with immunotherapy
KRAS G12C inhibitors including sotorasib and adagrasib have demonstrated clinical activity in the second line, with an improvement in response rate and PFS over docetaxel.62,63 However, sotorasib did not demonstrate a statistically significant improvement in OS over docetaxel. 63 Monotherapy use of a KRAS G12C inhibitor is unlikely to provide robust benefit in front line setting due to the resistance mechanism from KRAS G12C inhibition. 60 Therefore, multiple clinical trials were designed to assess the safety and efficacy of immunotherapy in combination with KRAS G12C inhibitors. As detailed above, there is additionally a strong preclinical rationale for combining a KRAS G12C inhibitor with immunotherapy. 94
Second line and beyond
Sotorasib is a small-molecule inhibitor of KRAS G12C that binds specifically and irreversibly. 63 In the second line, sotorasib demonstrated a response rate of 28.1% (95% CI: 21.5–35.4) and a mPFS of 5.6 months (95% CI: 4.3–7.8). 63 The first data presented the combination of immunotherapy with a KRAS inhibitor for patients with KRAS G12C mutations was from the CodeBreaK 100/101 studies, in which patients with KRAS G12C-mutated NSCLC received sotorasib at varying dose levels in combination with either atezolizumab 1200 mg q3w or pembrolizumab 200 mg q3w (see Table 1). Of patients enrolled, 67% had received prior immunotherapy, with a median of 1 prior line of therapy (range 0–7). Patients received sotorasib either concurrent with IO or sequentially with a sotorasib safety lead-in before starting IO. The combination was deemed unsafe to proceed with additional development, given that 57% (33/58) of patients had a grade 3 or 4 treatment-related adverse events, primarily hepatotoxicity. Despite the limiting toxicity, the combination showed that KRAS inhibitors plus immunotherapy could be efficacious, and lead to durable responses. The ORR was 29% (17/58), 83% disease control rate (48/58), and the duration of response was 17.9 months (95% CI: 5.6– not estimable (NE)). Median OS was 15.7 months (95% CI: 9.6–17.8). 95
KRAS G12C inhibitors in combination with immunotherapy.

AE, adverse event; KRAS G12Ci, Kirsten rat sarcoma G12C inhibitor; ORR, overall response rate; PD-L1, programmed death-ligand 1; RR, response rate; TRAE, treatment-related adverse event.
Olomorasib is a second-generation KRAS G12C inhibitor with significant single-agent activity and tolerability. 96 It has been studied in combination with pembrolizumab in 43 previously treated patients with NSCLC, of which 81% (35/43) had received prior immunotherapy. The ORR was 40% (17/43). The most common grade 3 toxicities observed were diarrhea (13%), ALT (6%), and AST (8%). 97 Data have not matured sufficiently to know the mPFS or mOS of this group.
Divarasib is a KRAS G12C inhibitor with high potency and selectivity that binds irreversibly in the inactive state. 98 Single-agent activity in a heavily pretreated patient population demonstrated an ORR was 53.4% (95% CI: 39.9–66.7), with an mPFS of 13.1 months (95% CI, 8.8 to could not be estimated). 98 Divarasib at a 200 and 400 mg dose has been combined with atezolizumab 1200 mg IV q3w in 39 patients who were previously treated, of which 90% (35/39) had received prior PD-1/PD-L1 inhibitor, 95% (37/39) received prior platinum chemotherapy, and 28% (11/39) had received a prior KRAS G12C inhibitor. The combination was safe, with no dose-limiting toxicities observed, and only 5% of grade 3 AST and ALT elevations were reversible. Confirmed ORR was 42.1% (n = 38), with an ORR of 55.6% (n = 27) for those without a prior KRAS G12C inhibitor.98,99 These trials taken together demonstrate that the combination of IO and KRAS G12C inhibition can be efficacious, though tolerability varies between KRAS G12C inhibitors.
Immunotherapy combinations in the first line
Given the clinical efficacy of KRAS G12C inhibitors and single-agent immunotherapy in the second line and beyond, there is significant interest in moving the combination up to the frontline. Adagrasib is a KRAS G12C inhibitor that binds irreversibly and selectively to KRAS in the inactive state. 3 As a single agent in the second line, it has an ORR of 32% with a PFS of 5.5 months, and a DOR of 8.3 months. 62 KRYSTAL-7 studied the efficacy and safety of adagrasib plus pembrolizumab in patients with treatment-naïve, advanced NSCLC with KRAS G12C mutations. Safety data were presented for 148 patients; while ALT and AST elevations were still frequently observed (38% and 32%, respectively), grade 3 elevations were uncommon (9% and 13%, respectively). Treatment discontinuations were limited, with discontinuation rates of adagrasib at 6% (9/148), pembrolizumab at 11% (16/148), and 4% discontinuing both (6/148). Of the 51 patients with PD-L1 ⩾50%, ORR was 63% (32/51, 95% CI: 48%–76%) with a disease control rate (DCR) of 84% (43/51; 95% CI: 71–93). Both median PFS and DOR were not reached with a median follow-up of 10.1 months. 100 Efficacy data for PD-L1 <50% have not been presented. Regarding future trials, for patients with TPS <50%, adagrasib will be studied in combination with pembrolizumab plus chemotherapy (ClinicalTrials.gov identifier: NCT05609578).
MK-1084 is a KRAS G12C inhibitor that binds covalently. 101 The combination of MK-1084 plus pembrolizumab 200 mg IV q3w was administered in patients with untreated, metastatic NSCLC with PD-L1 ⩾1%. Of the 27 patients who were response evaluable, the ORR was 70% (95% CI: 50%–86%) at MK-1084 doses ranging from 25 to 400 mg daily. Responses were seen both in patients who were PD-L1 1%–49% and PD-L1 ⩾50%. The combination was well tolerated, with grade 1/2 ALT elevation in 26% (8/31) and grade 3–5 in 13% (4/31); grade 1/2 AST elevation in 23% (7/31) and grade 3–5 in 10% (3/31). 102 The combination of MK-1084 plus pembrolizumab versus pembrolizumab plus placebo is being studied in a prospective, phase III trial of patients with KRAS G12C mutations and a PD-L1 ⩾50%. 103
Two cohorts of the LOXO-RAS-20001 study enrolled previously untreated patients with KRAS G12C to be treated with the combination of olomorasib plus pembrolizumab in the frontline setting, as well as olomorasib plus pembrolizumab plus chemotherapy. For patients treated with pembrolizumab plus olomorasib, there was an ORR of 77% (13/17). The data are not mature enough to be able to estimate PFS and OS. 97 The combination of olomorasib, pembrolizumab, and chemotherapy similarly showed a tolerable toxicity profile in 21 treatment-naïve patients. The ORR was 50%, with 48% (10/21) of patients having a PD-L1 <1%. 104 Both of these combinations will be pursued in the SUNRAY-01 trial. For patients who are PD-L1 ⩾50%, patients will be randomized to pembrolizumab plus olomorasib versus placebo. 97 Patients with any PD-L1 expression will be treated with platinum, pemetrexed, and pembrolizumab plus olomorasib versus placebo. 97
These data demonstrate that combinations of KRAS G12C inhibitors plus single-agent immunotherapy can be tolerable with significant clinical efficacy. We look forward to prospective, randomized data in the next few years. While outside the scope of this review on the role of immunotherapy in KRAS G12C NSCLC, the combinations of chemotherapy plus sotorasib will be studied in the randomized, phase III clinical trial CodeBreaK 202 in patients who are PD-L1 ⩽1%. 105 In addition, KRAS on-target inhibitors, pan-KARS inhibitors, or novel combination strategies may play a role in the treatment of KRAS G12C-mutated NSCLC. 106 It remains to be seen whether a single strategy will maximize efficacy, minimize toxicity, and increase CNS treatment activity, or whether an individualized approach based on patient characteristics, PD-L1, and co-mutations will be the best path forward.
Novel combinations with KRAS G12C inhibitors
As detailed above, there are multiple resistance mechanisms that have been identified after treatment with KRAS G12C inhibitors, including on-target resistance mechanisms, bypass mechanisms, and histologic transformation. 60 Currently, the genomic and histologic resistance profile following treatment with the combination of immunotherapy plus KRAS G12C inhibitors is unknown. Novel combinatorial strategies will be needed both to treat patients after the development of resistance to KRAS G12C inhibitors, as well as to prevent the initial development of resistance using novel combinations upfront. Many combination strategies with KRAS G12C inhibitors are under development, including the HER2 antibody–drug conjugate trastuzumab deruxtecan, a SOS1 inhibitor BI 1701963, the CDK4/6 inhibitor palbociclib, the PARP inhibitor olaparib, the EGFR inhibitor cetuximab, and the SHP2 inhibitor TNO155.107,108 Each of these combinations will have variable efficacy and toxicity, and may not be the correct treatment for an unselected, KRAS G12C-mutated population, but rather small subsets of patients. One aspirational goal is the treatment of every patient’s individual resistance mechanism with a combinatorial strategy that also minimizes toxicity, though much work needs to be done for this to be realized.
Conclusion
KRAS G12C inhibitors have demonstrated single-agent efficacy in later lines of therapy, although response rate and durability have been limited in randomized clinical trials. This has led to the investigation of immunotherapy-based strategies combined with KRAS G12C inhibition in the frontline, which have demonstrated promising response rates in multiple single-arm, phase I trials. The toxicity of KRAS G12C inhibitors appears drug-specific rather than a class effect, which has led to tolerability differences with unique combinations. It will be important to incorporate what we know about genomic and phenotypic changes with co-alterations to establish treatment paradigms in different subgroups such as STK11 or KEAP1 co-alterations. As the efficacy and tolerability of these combination strategies improve, moving them into earlier stages will be a viable strategy to improve survival for patients with KRAS G12C mutations.