
Abstract
Background:
Patients with unresectable upper gastrointestinal (UGI) cancers have limited treatment options and poor prognosis. Although phase I trials provide access to novel therapies, their benefits in this population are unclear.
Objectives:
We aimed to assess efficacy and survival outcomes of patients with refractory UGI cancers within phase I trials.
Design:
We conducted a retrospective pooled analysis of phase I trials enrolling patients with advanced UGI cancers who received at least one dose of the study drug at SCRI UK between 2011 and 2023.
Methods:
Efficacy and survival outcomes, including objective response rate (ORR), clinical benefit rate (CBR), disease control rate (DCR), duration of response, progression-free survival (PFS) and overall survival (OS), were assessed. Analyses were conducted for the entire cohort and stratified by trial agent class, molecularly matched therapy allocation and receipt of the recommended phase II dose (RP2D). Patients participating in multiple trials were analysed separately for each study.
Results:
From 1796 screened patients, 124 with UGI cancers were included in 37 phase I trials. Most were male (75%), with liver or peritoneal metastases (73%), treated with a median of 2 prior therapy lines. Of these, 60% received immunotherapy, 30% small molecules and 10% antibody-drug conjugates. Molecularly matched therapy was given to 22% and 86% received treatment at RP2D. In response-evaluable patients, ORR was 15%, CBR 40%, DCR 86% and median OS was 9.7 months. Treatment at RP2D was significantly associated with higher CBR (odds ratio 4.75, p = 0.04) and prolonged PFS (p = 0.04). Depth of response and treatment at RP2D were independent prognostic factors.
Conclusions:
Participation in phase I trials offers benefits in refractory upper gastrointestinal cancers with compelling results in late-line settings and potential early access to new therapies.
Background
Upper gastrointestinal (UGI; oesophageal/oesophagogastric junction/gastric) cancers have significant morbidity and mortality rates with an annual incidence of 9272 (oesophageal) and 6453 (gastric) per 100,000 UK population. Patients often present with stage IV disease and 1-year survival rates following diagnosis in this context remain extremely poor. 1
In recent years, the advent of monoclonal antibodies targeting the programmed death-(ligand)1 (PD-1/PD-L1) axis has changed the treatment paradigm of UGI tumours. Survival has extended, particularly for patients with PD-L1-expressing tumours.2–7 Furthermore, combinations of anti-PD(L)1 agent with other T-cell checkpoint modulators, for example, anti-T-cell immunoreceptor with Ig and ITIM domains (TIGIT) hold the potential to further improve clinical outcomes. 8
In contrast, targeting of oncogenic pathways has largely been unsuccessful in these cancers, with the exception of the anti-human epidermal growth factor receptor 2 (HER2) trastuzumab and the anti-vascular endothelial growth factor receptor 2 (VEGFR2) ramucirumab.9,10 Several studies have demonstrated the presence of significant tumour genomic heterogeneity, impairing the ability to identify clinically relevant molecular subgroups.11–13 As our understanding of tumour biology improves, increased numbers of successful targets and novel therapies are being identified, for example zolbetuximab in Claudin 18.2-expressing and bemarituzumab in FGFR2b-selected gastric or gastro-oesophageal junctional adenocarcinoma.14–16 Moreover, the development of antibody-drug conjugates (ADCs) targeting several molecules, including HER-2, Claudin 18.2, guanylyl cyclase C and trophoblast cell surface antigen 2 (Trop-2) may further ameliorate the prognosis in this population. 17
The majority of patients with UGI cancers have limited responses to standard lines of treatment. As a greater comprehension of tumour biology unveils increased numbers of possible therapeutic targets, the rates of patients with UGI cancers participating in phase I clinical trials are likely to increase. The key aims of phase I trials are to establish the recommended dose, schedule and toxicity profile of experimental drugs for subsequent disease-specific study in dedicated dose expansion cohorts and phase II trials. Such trials may offer an additional therapeutic option for a proportion of patients who remain fit despite having received multiple lines of conventional treatment.18–21
However, participation in clinical trials requires substantial patient selection, even more so in phase I trials that are generally confined to the treatment-refractory setting. Patients with UGI cancers often experience a significant number of symptoms limiting their performance status, which can preclude their eligibility for further treatments. As such, access to phase I trials has been traditionally limited with unclear benefits for patients with UGI cancers.
Sarah Cannon Research Institute United Kingdom (SCRI UK) is a leading clinical trials institution specialising in the development of new oncological therapeutic drugs. We conducted a retrospective review of all patients with metastatic or unresectable UGI cancers participating in phase I clinical trials at SCRI UK to assess disease-specific clinical outcomes.
Methods
We reviewed electronic medical records for patients with unresectable or metastatic UGI cancers enrolled in phase I trials at SCRI UK between November 2011 and February 2023. Patients were included if they qualified for trial enrolment and had received at least one dose of the trial drug. Patients who did not meet the eligibility criteria for trial participation or were withdrawn for other reasons before starting the trial treatment were excluded. All patients provided written informed consent for trial participation in accordance with the Declaration of Helsinki. Ethics approval was not required for this retrospective analysis as it involved the review of anonymised electronic medical records without any additional intervention or data collection being performed.
Patient demographics, tumour characteristics, prior treatments and trial therapy were collected from the electronic medical records for each patient.
Patient-level outcomes were extracted to estimate objective response rate (ORR; i.e., rates of subjects achieving complete response (CR) or partial response (PR)), clinical benefit rate (CBR; i.e., rates of subjects with any degree of tumour shrinkage) and disease control rate (DCR; rates of subjects achieving CR, PR or stable disease (SD)). ORR, CBR, DCR were assessed by site investigators according to Response Evaluation Criteria in Solid Tumours (RECIST) version 1.1 criteria. Time-to-event outcomes (progression-free survival (PFS), overall survival (OS), duration of response (DOR), duration of clinical benefit (DoCB)) were also determined for each patient. PFS was defined as the time from the commencement of each trial treatment to progression or death, whichever came first. OS was defined as the time from the start of trial treatment to death. DOR and DoCB were defined as the time from the commencement of each trial treatment to disease progression or death for the subjects achieving CRs/PRs or clinical benefit, respectively.
Subgroups analyses
The trial therapy was categorised according to the mechanism of action of the experimental arm which included small molecule inhibitors (SMs), immunotherapies (IOs) and ADCs. SMs were further subclassified into RAS/RAF pathway inhibitors (including BRAF, MEK, PI3K, MEK/FAK and RAS inhibitors), DNA damage response and repair (DDR) pathway inhibitors (ataxia telangiectasia and Rad3 related (ATR) inhibitors, poly (ADP-ribose) polymerase (PARP) inhibitors) and others (groups too small to be considered individually). IO included immune checkpoint inhibitors alone or in combination with anti-VEGF agents (IO-VEGF) and others (groups too small to be considered individually). In addition, the trial therapy was classified as molecularly matched if the enrolled patients were target-selected at study entry. Patients receiving the trial therapy at a dose that was equivalent to or higher than the established recommended phase II doses (RP2D) were considered treated at RP2D. For each of these groups, the efficacy outcomes were evaluated separately.
Statistical analyses
Descriptive statistics were used to summarise patient characteristics. Outcomes of patients enrolled in multiple phase I trials were assessed independently in each subsequent trial. These patients were only considered once in the assessment of patient demographics. Fisher’s exact test was used to assess the association of patient, tumour or trial characteristics with the efficacy outcomes. The Kaplan–Meier method was used to estimate time-to-event endpoints, with log-rank test used to compare survival curves and Cox regression models to estimate the hazard ratio (HR). Clinically valuable variables were selected for multivariable Cox regression model using a stepwise approach, with an entry criterion of p < 0.05 and a removal criterion of p > 0.10, as previously described. 22 Patients who did not experience the event of interest in the timeframe of our analysis were censored at the date of their last follow-up. Two-sided p-value <0.05 was set as the threshold for statistical significance.
All analyses were undertaken using IBM SPSS Statistics (IBM Corp. (2023). IBM SPSS Statistics for Windows (Version 29.0) [Computer software]. IBM Corp.) and R version 4.3.0 (R Core Team (2023). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria). The study complied with the relevant Equator network guideline. 23
Results
After screening a cohort of 1796 subjects, a total of 104 patients enrolled across 37 phase I clinical trials between November 2011 and February 2023 met eligibility criteria for assessment. Of these, 20 patients were enrolled in more than one trial, totalling a cohort of 124 trial subjects. The maximum number of trials that a single patient was enrolled in was 2. Most of the population comprised male patients (75%), with oesophageal/junction tumours (65%) and metastatic disease at diagnosis (73%). Of note, 73% of patients had either liver or peritoneal metastatic involvement. The median number of prior lines of systemic treatment received was 2 (range: 1–4). Detailed baseline patient and tumour characteristics are summarised in Table 1. A total of 100 patients (81%) had at least one on-trial tumour assessment and were deemed suitable for response assessment (response-evaluable cohort thereafter).
Baseline characteristics at trial entry.

ECOG PS, Eastern Cooperative Oncology Group Performance Status; GOJ, gastroesophageal junction; HER2, human epidermal growth factor receptor 2; n/a, not available.
HER-2 status was available for 83% of the patients (n = 86/104) and was positive in 17% (n = 18/104) of cases, in keeping with prior literature. 24 Microsatellite stability was evaluated in 45% (n = 47/104) of the cohort, and microsatellite instability (MSI) was reported in three cases (6.7%). Tumour PD-L1 assessment was only available in 14% (n = 15/104) of the cases and was therefore not included among the assessed variables.
A total of 38 distinct agents were tested across the 37 clinical trials eligible for this study. Thirty-seven patients (30%) received small molecule inhibitors, of which 15% were treated with agents targeting the RAS-MAPK pathway, 13% with drugs targeting the DDR genes and 2% with other SM therapies. A total of 74 patients (60%) received IO; these included single-agent anti-PD-(L)1 treatment (13%), anti-TIGIT (16%) with (n = 1) or without (n = 19) anti-PD-(L)1 inhibitor, and combinations of anti-PD(L)1 and anti-VEGFR treatments (28%). A small number (3%) of patients received other immunotherapeutic drugs including T-cell receptor targeting agents. Thirteen patients (10%) received an ADC. Within the entire cohort, 22% of patients (n = 27/124) received therapies for corresponding molecular alterations. These included aberrations in the RTK (41%, n = 11/27), DDR genes (52%, n = 14/27) or other intracellular (7%, n = 2/27) pathways. Among the subjects enrolled in IO- or ADC-testing trials, none was prospectively target-selected. Notably, all patients treated with molecularly matched therapies received monotherapies and, compared to the target-unselected cohort, had a higher number of metastatic sites (⩾2 in 41% vs 22%) and higher rates of liver and/or peritoneal disease (78% vs 57%). Baseline characteristics of the population according to their treatment allocation are provided in Supplemental Table 1.
In the response-evaluable cohort (n = 100) across all trials, the ORR was 15%, with no CRs and 15 PRs as per RECIST v1.1 criteria. CBR was 40% and DCR 86% (Table 2). The waterfall plot shows the best overall response for each evaluable patient (Figure 1(a)). Responses were similar by trial agent class (SM 14%, IO 11%, ADC 15%), even after the exclusion of the patients with known MSI-high tumours from the IO cohort (ORR 13%). A total of 86 patients were treated at RP2D in relatively similar proportions across trial categories (82% SM, 87% IO, 91% ADC). Responses were enriched in the subgroup treated at RP2D compared to their counterpart (ORR 16% vs 7%), and receiving treatment at RP2D was significantly associated with higher CBR (odds ratio (OR) 4.75, p = 0.04). Despite targeted treatment allocation not being significantly associated with either improved ORR or CBR in our cohort, responses were also enriched among patients receiving molecularly unmatched treatments (ORR 13% vs 7%), possibly reflecting the differences in baseline clinical and trial characteristics between molecularly matched and unmatched groups. None of the other patient, tumour or trial characteristics evaluated were significantly associated with either ORR or CBR (Supplemental Table 2). However, for combinatorial approaches targeting the PD(L)1-VEGF axis and drugs interacting with the DDR genes, which yielded the highest ORR (18%) and CBR (50%) respectively, there was a trend towards increased CBR (OR 0.43, p = 0.064).
Summary of efficacy outcomes.

Summary of the response outcomes in the entire population treated with at least a dose of the trial drug, in the cohort who had at least one on-trial restaging assessment (response-evaluable) and by clinically relevant subgroups.
No CRs were reported.
Outcomes of patients without known microsatellite instability (MSI-H) status in IO trials (n = 71), no. (%): ORR 9 (13); CBR 23 (32); DCR 51 (72).
Response outcomes by tumour characteristics imply that subjects enrolled in more than one trial are assessed independently.
ADC, antibody-drug conjugates; ATM, Ataxia Telangiectasia Mutated; CBR, clinical benefit rate; CR, complete response; DCR, disease control rate; ECOG PS, Eastern Cooperative Oncology Group Performance Status; IO, immunotherapy; MAPK, mitogen-activated protein kinase; ORR, objective response rate; PARP, poly-ADP ribose polymerase; PD(L)1, programmed death (ligand) 1; PR, partial response; RAS, Rat sarcoma virus; SD, stable disease; SMs, small molecules; TIGIT, T-cell immunoreceptor with Ig and ITIM domains; VEGFR, vascular endothelial growth factor receptor.
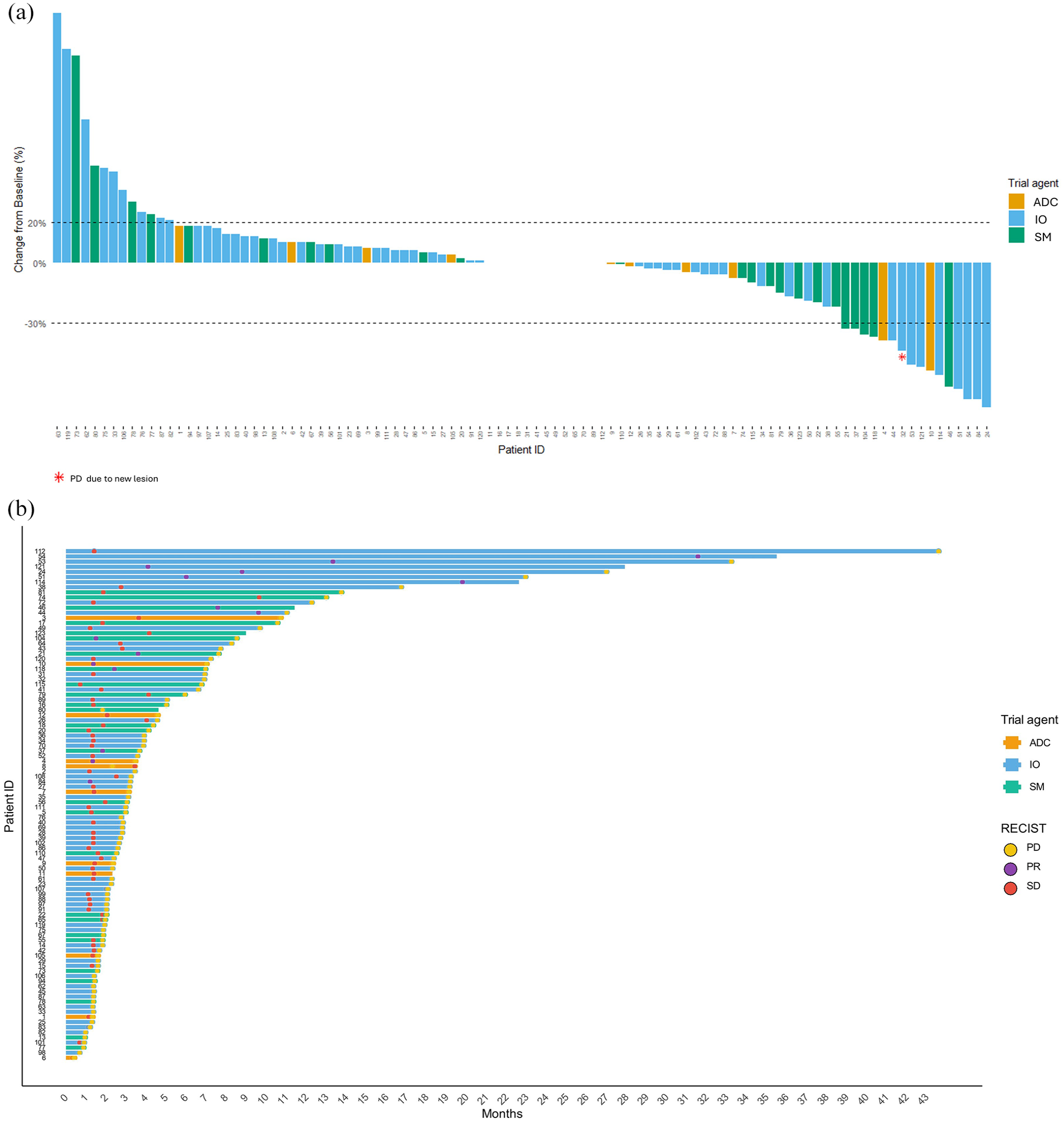
(a) Best overall response in patients with UGI cancers treated within phase I trials. (b) Duration of response in patients with UGI cancers treated within phase I trials.
Importantly, clinical benefit was observed regardless of age, location of primary tumour or number of previous lines of treatment that a patient had received. Clinical responses were also observed in prognostically unfavourable subgroups such as those with liver and/or peritoneal involvement.
Median follow-up across all trials was 14.79 months (95% CI, 3.63–25.95). Median OS was 8.02 months (95% CI, 6.03–10.02) in the overall population and 9.70 months (95% CI, 5.19–14.20) in the response-evaluable cohort. Median PFS was 2.83 months (95% CI 2.27–3.39) in the overall population, with a 6- and 12-month PFS rate of 25% and 10%, respectively. Among patients obtaining a radiological objective response (CR or PR) or clinical benefit from the trial treatment, median DOR and DoCB were 11.05 (95% CI, 3.68–33.27) and 5.95 months (95% CI, 3.48–8.28), respectively (Figure 1(b)). Notably, median DOR was longer with IO trial drugs (27.02 months, 95% CI, 3.22–33.27) compared to SMs (7.63, 95% CI, 3.68–8.55) or ADC (3.48, 95% CI, 3.48–7.04) despite the difference not reaching statistical significance (p = 0.06). Median DoCB was significantly longer in patients with Eastern Cooperative Oncology Group Performance Status (ECOG PS) 0 (p = 0.01). Further associations of clinicopathological and trial-related variables with DOR/DoCB are shown in Supplemental Table 3.
Achieving a radiological objective response (either CR or PR) within the phase I trial was associated with significantly longer median PFS and OS with durable benefit over time (Figure 2(a) and (b)). Additional prognostic factors for PFS and OS in the univariate model were obtaining any degree of tumour shrinkage with the trial drug and having a lower tumour burden at trial entry with less than two organs involved (Supplemental Figure 1(A) and (B) and Table 3(b)). Having a lower ECOG PS was also linked to improved PFS (p = 0.04). Of note, primary tumour site (oesophageal/junction vs gastric) and location of metastases (liver and/or peritoneum vs others) were not significant prognostic factors in our analyses. The main trial drug class (SM, IO or ADC) or receiving molecularly matched or unmatched treatments were not associated with a differential survival benefit either. However, treatments targeting the anti-PD(L)1/VEGF axis or the DDR pathway, which provided the highest ORR/CBR benefit in our cohort, yielded significantly longer OS (p = 0.028). Lastly, receiving treatment at RP2D was associated with longer PFS in both uni- (p = 0.035) and multivariable models (p = 0.018). The depth of radiological response (i.e. the percentage change in the sum of target lesions from baseline evaluated as continuous variable) achieved within the phase I study remained the strongest independent predictor of both PFS (HR 12.82, 95% CI, 6.48–25.36, p < 0.001) and OS (HR 3.54, 95% CI, 1.74–7.23, p < 0.001) in a multivariable model (Table 3(c) and Supplemental Figure 1).

Survival outcomes by best overall response in patients with UGI cancers treated within phase I trials. PFS (a) and OS (b) according to the best overall response achieved in the response-evaluable (n = 100/124) population estimated using the Kaplan–Meier method. Responders comprise PR; Non-responders SD and PD. Curves compared with log-rank test. Dashed area represents 95% CI.
Overall, uni- and multivariable survival analyses.

Statistically significant results are highlighted in bold.
ADC, antibody-drug conjugate; CI, confidence interval; ECOG PS, Eastern Cooperative Oncology Group Performance Status; HR, hazard ratio; IO, immunotherapy; No, number; OS, overall survival; PFS, progression-free survival; RP2D, recommended phase II dose; SM, small molecule.
Discussion
Patients with advanced UGI tumours have limited treatment opportunities and conventional late-line treatments result in survival times of 6–10 months, highlighting a critical need for novel, more effective treatments.10,25 However, participation in dose-escalation phase I studies has been traditionally challenging for patients with highly symptomatic and rapidly progressing cancers such as UGI tumours. Thus, whether enrolment in such studies grants patient-level benefits in this population has remained largely unknown. Our study demonstrates that patients with UGI cancers achieve meaningful clinical outcomes from participation in phase I trials. In our cohort, which primarily received the trial drugs as third-line treatment or beyond, ORR was 15%, DCR 86% and median OS was 8 months in the entire population and 9.7 months in the response-evaluable subgroup. These findings compare favourably to those reported for second-line and third-line standard-of-care agents and suggest that enrolment in phase I trials is valuable for these patients, offering an additional line of active treatment to some. Whilst we could not clarify what would be the optimal time for referral for phase I trials, early referral may be recommended given the observations that a more preserved performance status and a lower disease burden were associated with more favourable prognostic outcomes in univariable analyses in our study.
Phase I studies have now moved away from pure safety and dose-finding endpoints and increasingly include signal-seeking outcomes for accelerated drug development pathways. In our study, receiving treatment at RP2D was associated with higher clinical benefit and longer PFS and radiological responses (ORR and CBR) with improved PFS and OS. Importantly, the depth of radiological response qualified as the strongest independent predictor of improved PFS and OS in a multivariable analysis, suggesting that tumour shrinkage achieved in phase I settings is linked to survival even outside RECIST criteria. These findings suggest that modern phase I trials, particularly those incorporating disease-specific expansion cohorts, can provide a critical early efficacy appraisal of trial drugs for UGI cancers, accelerating therapeutic development and patient access to innovative treatments in this hard-to-treat disease.
Despite acknowledging patient selection in the context of early phase studies, it is worth noting that the majority of our trial patients had unfavourable prognostic factors, such as the primary tumour in situ (80%), metastases in ⩾2 organs (66%) and high rates of liver or peritoneal involvement (73%) at trial entry. Yet, clinical responses were achieved also in these subgroups. For instance, although the ORR was lower in patients with peritoneal disease, a finding which may also be accounted for by difficulties with the radiological assessment of these lesions, the DOR was similar across patients with or without peritoneal disease. Further, even though younger patients may be regarded as advantaged due to lower rates of comorbidities and a more preserved performance status, response rates and survival outcomes were not affected by age in our study. Lastly, overlapping clinical outcomes for gastric and oesophageal adenocarcinoma in our study align with the genomic similarities seen between these diseases and support uncoupled evaluation of the latter from oesophageal squamous cell carcinoma. 13
Interestingly, we did not observe superior outcomes in patients allocated to molecularly matched treatments, possibly due to the small cohort size, differences in baseline characteristics and variability in trial drug dosing and study designs. Notably, our molecularly matched subgroup had higher rates of unfavourable prognostic factors (e.g. higher tumour burden and metastatic liver and/or peritoneal involvement) and none received combination therapies compared to the target-unselected cohort. Phase I trials serve as a preliminary screening platform and may fail to confirm the relevance of putative targets identified through pre-clinical models. However, using adequate biomarker testing and allocation strategies and robust study designs are also instrumental to the success of precision medicine trials. Advances in molecular screening techniques, improved patient-matching platforms, including the widescale implementation of molecular tumour boards and the development of adaptive trial designs with multi-biomarker and multi-drug arms, may now provide a greater window of opportunity for accessing effective target-driven therapies in early phase studies. 26
Approaches to restore the endogenous anti-tumoural immune response by targeting angiogenesis and genomic instability may represent promising strategies in UGI cancers, as suggested by the significant OS improvement and trend towards better PFS and response outcomes seen with IO-VEGF or DDR-targeted agents in our study. In other solid tumours, targeting these pathways is a standard treatment option.27–31 In UGI tumours, VEGF-dependent pathways foster effector T-cell exhaustion and IO-VEGF combinations have shown promise in early-stage studies.32–34 Moreover, alterations in the homologous recombination repair system, especially Ataxia Telangiectasia Mutated (ATM) mutations, have been described in 7%–23% of the cases.35,36 This concept – together with the development of selective ATR inhibitors effective against PARP-resistant clones – reignites interest in synthetic lethality-based approaches (NCT04535401, NCT04657068) following disappointing results with PARP-inhbitors. 37
There are a number of limitations to take into account when interpreting the data presented. The study population comprised a heterogenous cohort of patients receiving a range of novel therapeutics in a late-line setting with the absence of a control group. Moreover, given the nature of phase I studies, variables are the doses of drugs administered across patients in different dose-level cohorts and in the number of lines of previous therapy. However, novel phase I trials have become better suited for drug efficacy assessments with the use of dose expansion cohorts, adaptive statistical endpoints and randomised designs. In this context, our study provides a contemporary insight into the outcomes of patients with upper GI cancers enrolled in phase I studies and supports the early referral and recruitment into such studies as these could offer compelling outcomes compared to the available late-line conventional therapies.
Supplemental Material
sj-docx-1-tam-10.1177_17588359251318864 – Supplemental material for Outcomes of patients with refractory upper GI cancers enrolled in phase I trials: a 10-year analysis from the Sarah Cannon Research Institute UK Drug Development Unit
Supplemental material, sj-docx-1-tam-10.1177_17588359251318864 for Outcomes of patients with refractory upper GI cancers enrolled in phase I trials: a 10-year analysis from the Sarah Cannon Research Institute UK Drug Development Unit by Cammarota Antonella, Joshi Kroopa, Aghayeva Farah, Woodford Rachel, Grochot Rafael, Williams Anja, Smyth Elizabeth Catherine and Fontana Elisa in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-2-tam-10.1177_17588359251318864 – Supplemental material for Outcomes of patients with refractory upper GI cancers enrolled in phase I trials: a 10-year analysis from the Sarah Cannon Research Institute UK Drug Development Unit
Supplemental material, sj-docx-2-tam-10.1177_17588359251318864 for Outcomes of patients with refractory upper GI cancers enrolled in phase I trials: a 10-year analysis from the Sarah Cannon Research Institute UK Drug Development Unit by Cammarota Antonella, Joshi Kroopa, Aghayeva Farah, Woodford Rachel, Grochot Rafael, Williams Anja, Smyth Elizabeth Catherine and Fontana Elisa in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
The authors would like to thank all the patients who participated in these studies. The funders had no role in the design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review or approval of the manuscript; and decision to submit manuscript for publication. The interpretation and reporting of these data are the sole responsibility of the authors.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.