
Abstract
Background:
Selinexor (SEL) is an oral inhibitor of nuclear export protein Exportin 1 (XPO1) previously shown to upregulate programmed cell death protein 1 (PD-L1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) expression.
Objective:
To investigate the safety and antitumor activity of SEL, nivolumab (NIVO), and ipilimumab (IPI) in Asian patients with treatment-refractory solid organ cancers.
Design:
Phase I study of escalating doses of SEL in combination with NIVO + IPI. Patients were enrolled in a 3 + 3 design.
Methods:
NIVO and IPI were dosed at 240 mg Q2W and 1 mg/kg Q6W, respectively. SEL was dosed with a 2-week monotherapy run-in prior to triplet therapy, at dose levels (DL) 1 (40 mg once/week) and DL2 (60 mg once/week). Dose-limiting toxicity (DLT) was assessed over the first 6 weeks.
Results:
Twelve patients were enrolled; 11 were evaluable for response, and 6 were evaluable for DLT (1 had a non-treatment-related stroke and 5 had progressive disease (PD) prior to completion of the DLT period). The median age was 64.5 (range 39–78) years. The median line of prior therapy was 3 (range 2–5). Dose escalation proceeded through DL1 (n = 7, 3 evaluable for DLT) and DL2 (n = 5, 3 evaluable for DLT). No DLTs were observed among evaluable patients. No patients required dose reduction of SEL. Most frequent treatment-related AEs were fatigue (5/12; G ⩾ 3 = 1), nausea (5/12; G ⩾ 3 = 0), anorexia (4/12; G ⩾ 3 = 0), transaminitis (2/12; G ⩾ 3 = 0), and hypomagnesemia (2/12; G ⩾ 3 = 0). The recommended phase II dose was SEL 60 mg once/week combined with NIVO + IPI. One patient had a partial response (PR; progression-free survival (PFS) of 61 days), 3 had prolonged stable disease (SD; PFS of 141, 344, and 442 days, respectively), and 7 had PD. All patients with SD or PR had previously progressed on immunotherapy but experienced prolonged disease control on SEL in combination with NIVO + IPI.
Conclusion:
SEL in combination with NIVO + IPI was well tolerated without any new safety signals. The combination showed promising and durable antitumor activity in Asian patients with advanced malignancies who had failed prior immunotherapy and merits further investigation.
Trial registration:
NCT04850755 (https://clinicaltrials.gov/study/NCT04850755).
Keywords
Introduction
Immunotherapy has had remarkable success in the treatment of cancer. However, many solid tumors still fail to respond to immune checkpoint inhibitors due to limited immunogenicity, unfavorable tumor microenvironments (TME), lack of infiltrating T lymphocytes, or increases in immunosuppressive Tregs. 1 A major focus now is to identify how to alter these characteristics, turning immunologically “cold” tumors into “hot” ones, to enhance the efficacy and broaden the applicability of immunotherapeutic strategies. 2
In cancers, tumor suppressor proteins (TSP) are rendered non-functional by being exported out of the nucleus by the nuclear export protein, Exportin 1 (XPO1), also previously known as chromosome region maintenance 1. Selinexor is a first-in-class, oral, slowly reversible, potent, and selective inhibitor of XPO1 that forces the nuclear retention and activation of multiple TSPs, restricting tumor progression.3–5 Restoration of TSP pathways has been shown to induce immunomodulatory activities through NAFTc1-STAT3 signal transduction.6,7
Preclinical studies have demonstrated that selinexor upregulates programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) expression and marginally suppresses programmed death-ligand 1 (PD-L1) expression in human leukocytes, ex vivo. 7 In studies assessing XPO1 inhibitors and CD19-directed CAR-T cells, pretreatment of target cells with an XPO1 inhibitor reduced the expression of T-cell exhaustion markers, including PD-1, LAG-3, and Tim-3.8,9 Selinexor has also been shown to induce PD-L1 gene expression by ~2-fold in B16F10 melanoma tumor cell lines in vitro. 10 In addition, synergistic effects of PD-1/PD-L1 inhibition with selinexor have been observed in preclinical solid tumor models such as melanoma 10 and renal cell carcinoma, 11 where concurrent selinexor administration altered the immune environment by increasing the number of T cells and NKT cells with a simultaneous reduction in Gr1+ and CD11b+ myeloid cells and FOXP3+ regulatory T cells.10,11 The combination of selinexor with PD-1 or PD-L1 blockade also showed significant immune-modulatory activity, inducing changes in the frequency and phenotype of immune cell populations, including increased frequency of NK cells, induced differentiated TH1 cells, and increased frequency of activated T cells in vitro. 10
While no preclinical studies have evaluated the triplet combination of selinexor (SEL), nivolumab (NIVO), and ipilimumab (IPI), two independent groups have investigated the combination of selinexor with either PD-1 or CTLA-4 blockade.7,11 In syngeneic melanoma mouse models, selinexor combined with anti-PD-1/PD-L1 antibodies led to reduced tumor burden and was associated with enhanced effector T-cell and NKT-cell infiltration and diminished immunosuppressive myeloid and regulatory T-cell populations. Similarly, co-administration of selinexor with anti-CTLA-4 antibodies resulted in tumor regression. 7 These responses are potentially mediated by decreased nuclear export of key immunomodulatory molecules, specifically STAT3 and NFATc1, which are involved in the regulation of immune checkpoints, thereby enhancing the efficacy of immune checkpoint inhibition. 12 These results collectively support the clinical evaluation of the combination of selinexor + PD-1/CTLA-4 blockade in solid tumors.
We conducted an investigator-initiated, single-center phase I study evaluating the safety and tolerability of selinexor in combination with nivolumab and ipilimumab. Primary objectives included safety and determination of the maximum tolerated dose (MTD) and recommended phase II dose (RP2D) of selinexor in combination with nivolumab and ipilimumab; secondary objectives included antitumor activity of this combination.
Methods
Patient population
NEXUS was an investigator-initiated, single-center, phase I clinical trial evaluating the safety and tolerability of selinexor in combination with nivolumab and ipilimumab in Asian patients with advanced, refractory solid tumors. The study was conducted in accordance with protocol requirements, the International Conference on Harmonization for Good Clinical Practice, and the guiding principles in the Declaration of Helsinki, according to local laws and regulations.
Eligible patients were recruited from the National University Cancer Institute, Singapore. Patients aged 21 years or older, with advanced solid tumors refractory to standard treatment, Eastern Cooperative Oncology Group performance status of 0 or 1, measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, 13 and adequate bone marrow, liver, kidney, coagulation, and cardiac function. Patients with previous disease progression on immunotherapy (including anti-PD-1/PD-L1 or anti-CTLA-4 antibodies) were eligible. Key exclusion criteria included radiation or systemic anticancer therapy ⩽3 weeks prior to the first dose of study treatment, uncontrolled active infection, known HIV infection, active autoimmune disease, and chronic treatment with systemic steroids equivalent to >10 mg/day prednisone. Topical, inhaled, nasal, ophthalmic steroids, and replacement-dose steroids for adrenal insufficiency were allowed. Patients who had prior grade 4 immune-related adverse events (AEs) from prior immunotherapy (including anti-PD-1/PD-L1 or anti-CTLA-4 antibodies) were excluded. Full eligibility criteria are provided in the trial protocol.
Trial design and interventions
NEXUS was a phase I trial investigating the safety, tolerability, and antitumor activity of escalating doses of selinexor in combination with nivolumab and ipilimumab. All patients received a 14-day run-in period of selinexor monotherapy prior to the first dose of nivolumab and ipilimumab. Ipilimumab was administered for a maximum of four cycles while nivolumab and selinexor were continued for up to 24 months in total or until discontinuation criteria were met (Figure 1).

Dosing schedule. Patients were treated with a 42-day schedule. Patients were dosed with selinexor once a week continuously on Days 1, 8, 15, 22, 29, and 36. Nivolumab was administered on D1, D15, and D29 of each cycle at 240 mg. Ipilimumab was dosed only on D1 of each cycle at 1 mg/kg. Ipilimumab was continued for a maximum of four cycles. Nivolumab and selinexor were continued for up to 24 months or until discontinuation criteria were met.
When the trial was designed in 2019, multiple dosing regimens for ipilimumab in combination with nivolumab were considered appropriate across various tumor types and indications. The phase I CheckMate 012 trial in non-small-cell lung cancer (NSCLC) assessed nivolumab at 3 mg/kg every 2 weeks, combined with ipilimumab at 1 mg/kg every 6 or 12 weeks. 14 The study indicated that these schedules had improved tolerability compared to others, as grade 3–4 treatment-related adverse events (TRAEs) occurred in 28% of patients; this is significantly lower than the 46%–48% seen with higher dose-intensity or dose-density regimens. In addition, treatment discontinuation due to AEs in patients who received ipilimumab at 1 mg/kg every 6 weeks was 13%, compared to up to 22%–33% in other dosing schedules.15,16 Furthermore, CheckMate 227 demonstrated that nivolumab combined with ipilimumab at 1 mg/kg every 6 weeks was superior to chemotherapy alone in patients with NSCLC, while maintaining a favorable safety profile. 17 Based on these data, we chose the ipilimumab 1 mg/kg every 6 weeks dosing regimen as it provided an optimal balance between efficacy and reduced toxicity.
All patients were treated until disease progression (as defined by RECIST 1.1 criteria) or intolerable toxicity. Disease assessment was performed every 6 weeks to assess response for the first four cycles, then 12 weekly thereafter. All AEs were recorded and graded using the National Cancer Institute Common Terminology Criteria for Adverse Events v 4.03 up to 30 days after the last dose. In the event where toxicity was attributable to only one agent, the agent causing toxicity was discontinued, but the patient was permitted to continue treatment with the other agents if there was evidence of ongoing clinical benefit.
A 3 + 3 dose escalation design was employed, beginning at dose level (DL) 1 (Table 1), with provisions for de-escalation if warranted by toxicity. A minimum of three patients were enrolled per cohort. Following the completion of at least one cycle (6 weeks) of triplet therapy at the target dose by the initial three patients, up to three additional patients could be enrolled in the same cohort. Each DL was closed to accrual pending a formal safety review of all patients (n = 3–6), conducted via a safety cohort meeting at the end of cycle 1. This meeting was convened by the site study team in conjunction with the medical monitor from Karyopharm. The study protocol was subject to oversight by both the Health Sciences Authority of Singapore and the National University Health System Research Office. If the DL was deemed tolerable over the 6-week observation period, escalation proceeded to the next DL. Dose interruptions or reductions were permitted and could be applied independently to each study drug based on the attribution of observed toxicities.
Dose escalation schedule for selinexor in combination with nivolumab and ipilimumab.

IV, intravenous; PO, peroral.
While a dose expansion phase was originally planned following dose escalation, it was not pursued as the study sponsor discontinued funding due to a change in strategic direction.
Study endpoints
Primary endpoints included safety and determination of MTD, dose-limiting toxicity (DLT), and RP2D. DLT were evaluable in patients who met the inclusion criteria on the first day of dosing with all three agents. Patients who passed screening but did not pass inclusion/exclusion criteria on day 1 of dosing of all three drugs were included in the study but are not evaluable for DLT. DLT was defined as any of the following occurring in the first 42 days at the target dose of study participation that was considered at least possibly related to drug administration: grade 3 nausea/vomiting or diarrhea lasting for more than 7 days while taking optimal supportive medications, grade 3 fatigue lasting for more than 5 days while taking optimal supportive care, grade 3 transaminitis lasting more than 7 days or occurring in the setting of bilirubin elevation more than two times upper limit normal, any other clinically significant grade 3 non-hematologic toxicity requiring intervention or admission, grade 4 neutropenia lasting more than 7 days, febrile neutropenia or grade 3 thrombocytopenia associated with bleeding, more than two missed doses in 42 days at the target dose due to a toxicity that was at least possibly study drug related, or any clinically significant occurrence that was at least possibly drug related in which the investigators and Sponsor agreed would place subjects at undue risk to their safety. Patients who missed more than two doses during Cycle 1 for reasons unrelated to the study drugs were considered not evaluable for DLT and were replaced.
Secondary endpoints included objective response rate (ORR) by RECIST 1.1, progression-free survival (PFS), overall survival (OS), and clinical benefit rate (CBR). CBR was defined as complete or partial response (PR), or stable disease (SD) lasting ⩾16 weeks per RECIST 1.1.
Nivolumab and ipilimumab are monoclonal antibodies that are degraded by proteolytic enzymes and cleared by the reticuloendothelial system, similar to endogenous IgG, 18 while selinexor is metabolized by CYP3A4, multiple UDP-glucuronosyltransferases, and glutathione S-transferases. 19 Due to the very low predicted risk of drug–drug interactions, we did not pursue pharmacokinetic exploration.
The reporting of this study conforms to the CONSORT-DEFINE statement 20 (Supplemental Appendix 1).
DNA sequencing
Comprehensive genomic profiling of formalin-fixed and paraffin embedded pre-treatment tumor samples was performed with the FoundationOne CDx assay (Foundation Medicine, Inc., Cambridge, Massachusetts, USA) for detection of substitutions, insertion and deletion alterations, and copy number alterations in 324 genes and select gene rearrangements and tumor mutational burden (TMB). Fresh biopsy was required at baseline, but if insufficient for next-generation sequencing, the trial permitted sequencing of archival tissue obtained within 1 year of enrollment. Analyses and data visualizations were done in R version 4.3.2 (R Core Team, 2024) the ComplexHeatmap R package (version 2.22.0).
Statistical analysis
A 3 + 3 dose escalation design was applied to determine the RP2D. PFS and OS were assessed with the Kaplan–Meier method. The median duration of follow-up was calculated by the reverse Kaplan–Meier approach. All statistical analyses pertaining to survival analyses and duration of follow-up were performed in R (version 4.3.2) using the survminer R package (version 0.5.0) and survival R package (version 3.8.3), and performed on an intention-to-treat (ITT) basis. For assessment of ORR and CBR, we performed analyses based on modified ITT to include only those with evaluable disease per RECIST 1.1.
Results
Patients characteristics
Twelve patients were enrolled and included in the safety analysis. The first patient was enrolled on April 1, 2021, and the final patient on August 18, 2022. Eleven were evaluable for response, and six were evaluable for DLT (one had a non-treatment related stroke and five had progressive disease (PD) prior to completion of the DLT period; Figure 2). The median age at enrollment was 64.5 (range 39–78) years, and the median line of prior therapy in the metastatic setting was 3 (range 2–5). The median duration of follow-up was 21.6 (95% confidence interval (CI): 19.3–24.5) months. Nine (75%) of the 12 patients enrolled had prior progression on immunotherapy. Additional baseline characteristics are summarized in Table 2.

CONSORT diagram.
Baseline characteristics.

PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; SCC, squamous cell carcinoma; TCC, transitional cell carcinoma.
Establishment of MTD and RP2D
A total of seven patients were enrolled at DL1, of whom three patients were evaluable for DLT. One patient developed a non-treatment-related stroke, and three patients developed PD prior to completion of the DLT period and were replaced. Of the three patients who completed the DLT period, none developed DLTs, and no patients required dose reduction of selinexor. Five patients were subsequently enrolled at DL2, and two patients developed disease progression during the DLT period and were replaced. Of the three patients who completed the DLT period at DL2, no DLTs were observed, and none of the patients required dose reduction of Selinexor. Thus, DL2 selinexor 60 mg once weekly in combination with nivolumab and ipilimumab was declared as the RP2D.
Safety and toxicity
The most frequent TRAEs were fatigue (5/12; Grade (G) ⩾ 3 = 1), nausea (5/12; G ⩾ 3 = 0), anorexia (4/12; G ⩾ 3 = 0), transaminitis (2/12; G ⩾ 3 = 0), and hypomagnesemia (2/12; G ⩾ 3 = 0; Table 3). The most frequent treatment emergent AEs were fatigue (6/12; G ⩾ 3 = 1), nausea (5/12; G ⩾ 3 = 0), anorexia (5/12; G ⩾ 3 = 0), fever (4/12; G ⩾ 3 = 4), anemia (2/12; G ⩾ 3 = 0), hypomagnesemia (2/12; G ⩾ 3 = 0), hyponatremia (2/12; G ⩾ 3 = 1), elevated lipase (2/12; G ⩾ 3 = 0), dyspnea (2/12; G ⩾ 3 = 1), stroke (2/12; G ⩾ 3 = 2), ascites (2/12; G ⩾ 3 = 2), and rash (2/12; G ⩾ 3 = 0; Table 4). Of note, few patients developed toxicities that were likely immune-related AEs—one patient developed dry eyes that was deemed to be immune-mediated. One patient passed away from fatal bleeding, which occurred in the context of tumor response—the patient had significant tumor shrinkage of her fungating groin tumor in response to the triplet regimen, but subsequently presented to the emergency department with torrential fatal bleeding from the underlying exposed femoral artery (Supplemental Figure 1).
Treatment-related AEs (n = 12).

AE, adverse event.
Treatment-emergent AEs (n = 12).

AE, adverse event.
Antitumor responses and survival analyses
Ten patients were evaluable for response. One patient had PR (PFS of 61 days), three patients had SD (PFS of 141, 344, and 442 days), and six patients had PD, corresponding to an ORR of 10% and CBR of 40%. Of note, all patients with SD or PR had previously received and progressed on immunotherapy (Table 5). Figure 3 shows the swimmer and waterfall plots of patients with evaluable disease. Figure 4 shows the mutational landscape at baseline, which was available for seven of the evaluable patients. The most common pathogenic alterations were in TP53, KRAS, MTAP, and TET2, present in 29% (two out of seven) of patients. TMB data were evaluable for five of the seven patients; two patients had high TMB defined as ⩾10 mutations/megabase.
Evaluable patients with PR or SD to treatment.

MS, micro-satellite; PFS, progression-free survival; PR, partial response; SD, stable disease.
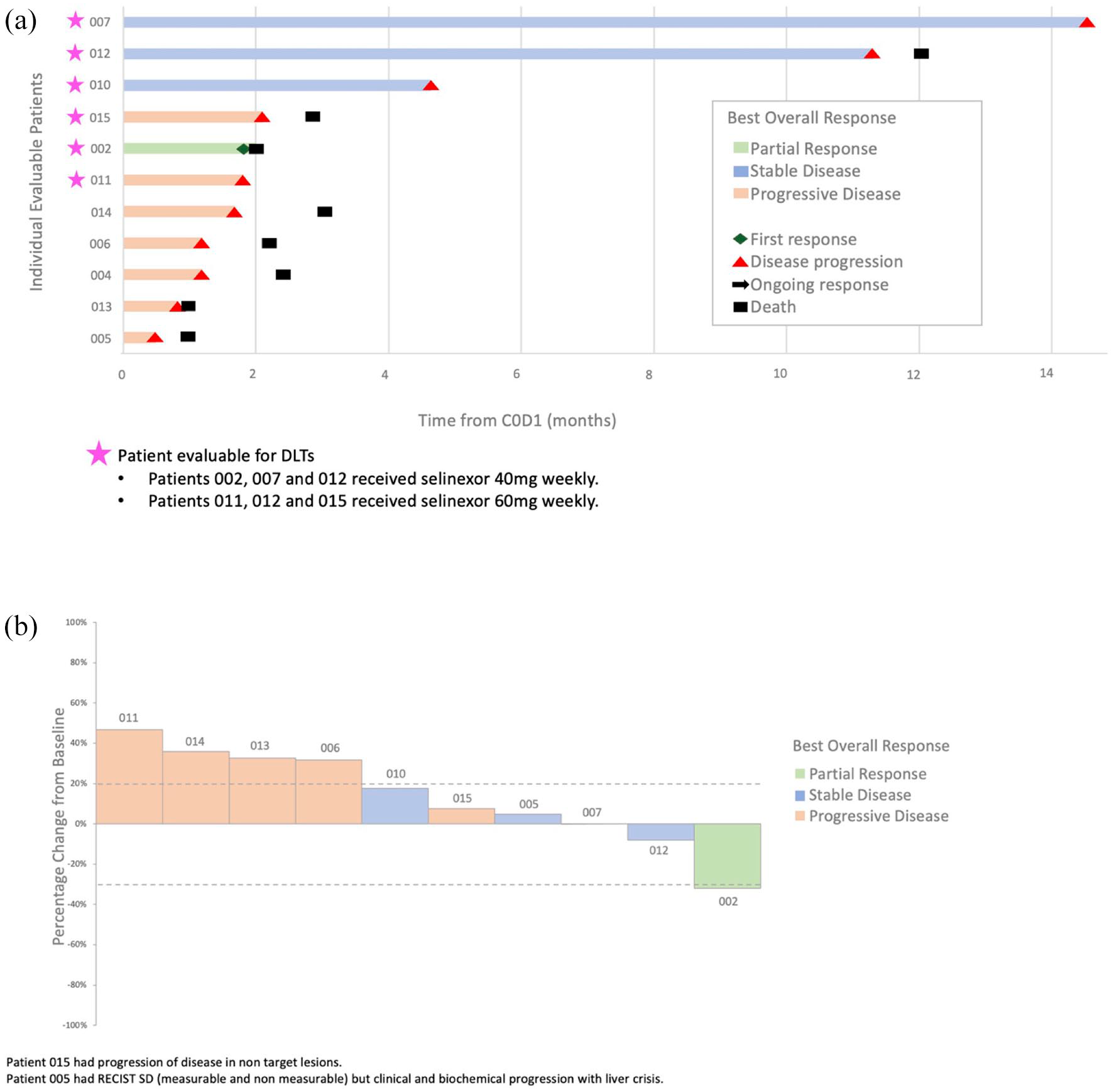
Clinical efficacy in patients with evaluable disease. (a) Swimmer plot of patients with evaluable disease. (b) Waterfall plot of patients with evaluable disease.
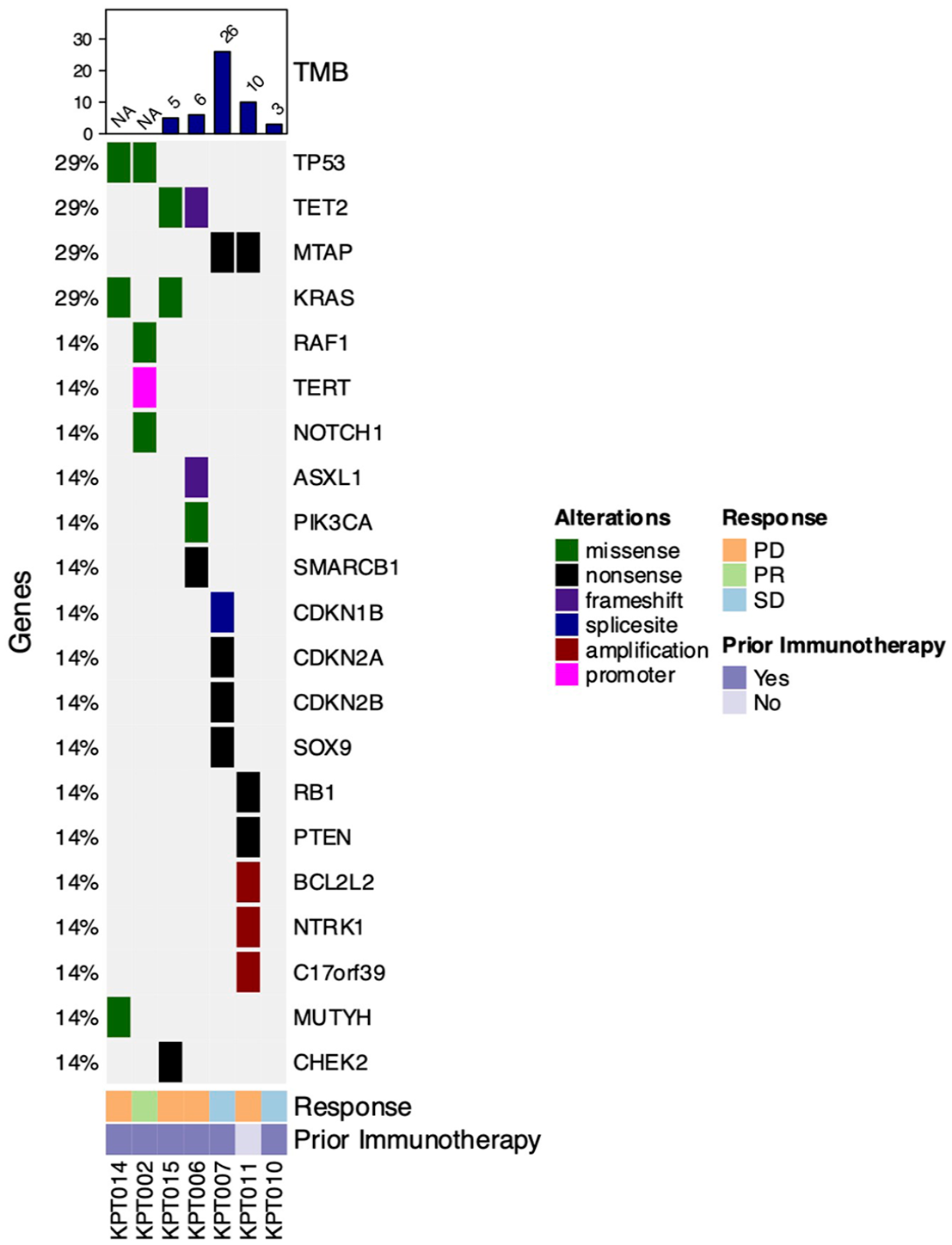
Mutational landscape at baseline. Heatmap demonstrating the pathogenic genomic alterations identified from most common to least common.
Discussion
In this investigator-initiated phase I trial evaluating the safety and tolerability of selinexor in combination with nivolumab and ipilimumab in patients with advanced refractory solid tumors, the combination was well tolerated with no new safety signals identified. The toxicity profile of the combination was manageable, consistent with that observed with selinexor monotherapy or ICIs. Few treatment-emergent immune-mediated toxicities were observed; there were no dose-limiting toxicities, and none of the patients required dose reduction of selinexor. The RP2D was selinexor 60 mg once weekly in combination with nivolumab and ipilimumab. We observed an ORR of 10% and a CBR of 40%. The triplet combination showed promising antitumor activity in Asian patients with advanced malignancies, including those who had failed prior immunotherapy with anti-PD-1 or anti-PD-L1 agents.
The favorable tolerability profile may in part be attributable to the use of low-dose ipilimumab (1 mg/kg every 6 weeks), a strategy that has been associated with reduced immune-related toxicity.14,21 While some data from melanoma studies suggest this dosing schedule may reduce efficacy, 22 updated findings from CheckMate 227 in NSCLC demonstrated numerically improved survival with low-dose ipilimumab plus nivolumab compared to nivolumab-containing arms across PD-L1 subgroups, supporting the notion that ipilimumab 1 mg/kg every 6 weeks maintains clinically effective CTLA-4 blockade. 23
Despite funding constraints limiting the expansion of dose cohorts, the early efficacy signals observed in our study suggest that this combination has the potential to re-sensitize tumors to checkpoint blockade. In a patient population where responses to further immunotherapy are typically poor, these early findings are clinically meaningful and warrant further evaluation. Globally, there is growing interest in strategies to overcome PD-1 resistance, including epigenetic modulators, TME-targeting agents, and multi-checkpoint blockade approaches.24,25 However, many of these efforts are still in early investigational stages, and no standard-of-care approach has been established for patients who progress on PD-1/PD-L1 inhibitors. The combination of selinexor, nivolumab, and ipilimumab is a scientifically rational, mechanistically novel, and clinically promising approach to overcoming resistance to PD-1 blockade that is also safe and well tolerated. Given the unmet need in this population, future studies are warranted to confirm therapeutic activity and to define the patient populations most likely to benefit. A prospective phase II study enrolling patients with documented progression on prior PD-1/PD-L1 therapy would allow further evaluation of this combination. Key objectives would include ORR, PFS, OS, and biomarker correlates of response.
Limitations of this trial include its small sample size and lack of a dose expansion phase to further explore the safety and efficacy signals seen in dose escalation. Furthermore, several patients had early disease progression within 2–4 weeks of commencing combination therapy. Preclinical studies have demonstrated that selinexor can induce PD-L1 gene expression and upregulate PD-1 and CTLA-4 expression. 7 Therefore, the study was designed with a selinexor monotherapy run-in phase for 2 weeks, to incorporate paired pre- and post-selinexor tumor biopsies for evaluation of tumor immune microenvironment changes. Due to procedural risks, paired tumor biopsies could only be safely performed in a limited number of patients, and the collected samples consisted of non-viable tissue, which was unsuitable for evaluation of treatment-induced changes. Given the limited activity of selinexor monotherapy in solid tumors (ORR 7%–8%26,27), the delay in initiating immunotherapy may have limited the opportunity for clinical benefit in patients with highly proliferative disease, resulting in progression prior to completion of the DLT evaluation period. Other groups have administered selinexor concurrently with ICI without a run-in phase,28–30 suggesting that this may be a promising strategy to reduce early disease progression. However, in patients with clinical benefit from the triplet combination in our study, durable disease control was observed, and, intriguingly, these occurred in patients who had failed prior anti-PD-1/PD-L1 therapy.
Previous studies have demonstrated that selinexor is safe in combination with single-agent anti-PD-1 blockade with meaningful clinical efficacy. In heavily pre-treated NSCLC patients, the combination of selinexor and pembrolizumab demonstrated an ORR of 18% and a 6-month disease control rate of 24%; half of the patients with durable disease control were those who had prior disease progression on immunotherapy. 28 Another small retrospective study of five NK T-cell lymphoma patients who had exhausted all treatment options, including treatment failure L-asparaginase regimens and prior anti-PD-1 blockade, showed remarkable responses to selinexor and tislelizumab, where 80% (four patients) achieved objective responses (three complete responses, one PR). 29 Similar to our findings, these data demonstrate that a subset of patients with prior acquired resistance to anti-PD-1 blockade may achieve durable disease control on ICI rechallenge when given in combination with selinexor. Furthermore, although it is impossible to tease out the exact contribution of each component within the triplet combination, these studies support the notion that selinexor plays a key role in re-sensitizing tumors to anti-PD-1 therapy.
In terms of mechanisms driving clinical benefit from selinexor and ICI, Bracht et al. 31 showed that HMGB1 mRNA appears to be a negative prognostic marker for immunotherapy response in lung cancer; they demonstrated that selinexor prevented nuclear export and release of HMGB1 from the cytoplasm in the PC9 lung cancer cell line and was able to inhibit cell proliferation in EGFR and KRAS mutant cell lines, suggesting that the combination of selinexor and ICI could yield better responses to immunotherapy. 31 Recently, Vergote et al. 32 evaluated the role of maintenance selinexor in patients with endometrial cancer who achieved partial or complete response with first-line taxane–platinum chemotherapy. In the primary analysis, there was no significant improvement in PFS; however, in the pre-specified subgroup analysis of TP53 wild-type patients, remarkable improvement in median PFS 20.8 versus 5.2 months (hazard ratio 0.46, p = 0.002) was seen at the latest updated long term analysis, 33 with efficacy seen regardless of microsatellite status. This is consistent with the mechanism of Selinexor, which drives nuclear retention and functional activation of wild-type, biologically active TSPs, including TP53. Similarly, the presence of TP53 mutations predicted weaker tumor response to selinexor in KRAS mutant lung adenocarcinoma patient-derived xenografts. 34 These studies highlight the importance of identifying biomarkers of response to select candidates who will benefit from treatment incorporating selinexor. Of note, the presence of TP53 mutations, in particular the R175H mutation, has also been shown to predict for lack of response to immunotherapy. 35 We performed baseline next-generation sequencing for 7 of the 11 evaluable patients; 2 patients had TP53 mutations present at baseline, 1 of the patients had a PR to the triplet combination, and 1 had PD. In our study cohort, the presence of TP53 mutations did not appear to be associated with a lack of response to selinexor and ICI therapy, suggesting that other mechanisms of response are at play. Further studies are needed to identify patients who benefit from the combination of selinexor and ICI.
Conclusion
Selinexor in combination with nivolumab and ipilimumab was well tolerated without any unexpected safety signals. The combination showed promising antitumor activity in Asian patients with advanced malignancies who had failed prior immunotherapy and merits further investigation.
Supplemental Material
sj-docx-1-tam-10.1177_17588359251339930 – Supplemental material for NEXUS: a phase I dose escalation study of selinexor plus nivolumab and ipilimumab in Asian patients with advanced/metastatic solid malignancies
Supplemental material, sj-docx-1-tam-10.1177_17588359251339930 for NEXUS: a phase I dose escalation study of selinexor plus nivolumab and ipilimumab in Asian patients with advanced/metastatic solid malignancies by Joan R. Choo, Santhiay Nathan Jeraj, Raghav Sundar, Wei Peng Yong, Matilda Lee, Yiqing Huang, Yugarajah Asokumaran, Gloria H. J. Chan, Natalie Y. L. Ngoi, Andrea L. A. Wong, Ross A. Soo, Cheng Ean Chee, Joline S. J. Lim, Boon Cher Goh, Soo Chin Lee and David S. P. Tan in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-2-tam-10.1177_17588359251339930 – Supplemental material for NEXUS: a phase I dose escalation study of selinexor plus nivolumab and ipilimumab in Asian patients with advanced/metastatic solid malignancies
Supplemental material, sj-docx-2-tam-10.1177_17588359251339930 for NEXUS: a phase I dose escalation study of selinexor plus nivolumab and ipilimumab in Asian patients with advanced/metastatic solid malignancies by Joan R. Choo, Santhiay Nathan Jeraj, Raghav Sundar, Wei Peng Yong, Matilda Lee, Yiqing Huang, Yugarajah Asokumaran, Gloria H. J. Chan, Natalie Y. L. Ngoi, Andrea L. A. Wong, Ross A. Soo, Cheng Ean Chee, Joline S. J. Lim, Boon Cher Goh, Soo Chin Lee and David S. P. Tan in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-3-tam-10.1177_17588359251339930 – Supplemental material for NEXUS: a phase I dose escalation study of selinexor plus nivolumab and ipilimumab in Asian patients with advanced/metastatic solid malignancies
Supplemental material, sj-docx-3-tam-10.1177_17588359251339930 for NEXUS: a phase I dose escalation study of selinexor plus nivolumab and ipilimumab in Asian patients with advanced/metastatic solid malignancies by Joan R. Choo, Santhiay Nathan Jeraj, Raghav Sundar, Wei Peng Yong, Matilda Lee, Yiqing Huang, Yugarajah Asokumaran, Gloria H. J. Chan, Natalie Y. L. Ngoi, Andrea L. A. Wong, Ross A. Soo, Cheng Ean Chee, Joline S. J. Lim, Boon Cher Goh, Soo Chin Lee and David S. P. Tan in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
This study was supported by grant funding from Karyopharm Therapeutics and the National Medical Research Council (NMRC) Clinician Scientist Award Senior Investigator Grant (CSASI21jun-0003) to D.S.P.T. Study drug Selinexor was provided by Karyopharm Therapeutics, while study drugs nivolumab and ipilimumab were provided by Bristol Myers Squibb. We are thankful to all patients who participated in this study. Preliminary findings from this study were presented at the ESMO Asia Congress 2024, European Society for Medical Oncology, December 6–8, Singapore.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.