
Abstract
The enhancer of zeste homolog 2 (EZH2) is a catalytic component of Polycomb repressive complex 2 (PRC2) mediating the methylation of histone 3 lysine 27 (H3K27me3) and hence the epigenetic repression of target genes, known as canonical function. Growing evidence indicates that EZH2 has non-canonical roles that are exerted as PRC2-dependent and PRC2-independent methylation of non-histone proteins, and methyltransferase-independent interactions of EZH2 with various proteins contributing to gene expression regulation and alterations in the protein stability. EZH2 is frequently mutated and/or its expression is deregulated in various cancer types. The cancer sensitivity to inhibitors of EZH2 enzymatic activity and state-of-the-art approaches to deplete EZH2 with chemical degraders are discussed. This review also presents the clinical trials in various phases that evaluate the use of EZH2 inhibitors, both as monotherapy and in combination with other agents for the treatment of patients with diverse types of cancers.
Background
Enhancer of zeste homolog 2 (EZH2) is one of the epigenetic modifiers that contribute to the global and focal transcriptional output, thus defining normal organismal development and developmental disorders, as well as cancer and mental health diseases. 1 It is the catalytic subunit of Polycomb repressive complex 2 (PRC2), a protein complex that contains also embryonic ectoderm development (EED), suppressor of zeste 12 (SUZ12), and retinoblastoma cell tumor-associated proteins 46 and 48 (RbAp46/48).1,2 In addition to these core components, several facultative protein subunits such as AE-binding protein 2 (AEBP2), jumonji and AT-rich interaction domain containing 2 (JARID2), Polycomb-like (PCL) proteins, and various RNA molecules can interact with PRC2 defining the specificity of its binding to chromatin and activity.1,3–5 PRC2 mediates mono-, di-, and trimethylation of the lysine 27 on the tail of histone 3 (H3K27me1/2/3) (Figure 1). While H3K27me1 is found in actively transcribed loci, H3K27me3 is associated with chromatin compaction and epigenetic silencing. 6 The transfer of a methyl group from S-adenosyl methionine (SAM) to H3K27 is catalyzed by an enzymatic domain of EZH2 named SET (Su(var)3-9, enhancer of zeste, and Trithorax). SET domain is not active unless EZH2 interacts with SUZ12 and EED.7,8

Schematic presentation of canonical and non-canonical function of EZH2. Canonically, EZH2 as an enzymatic subunit of PRC2 silences target genes by trimethylation of H3K27. Several facultative protein subunits, such as PCL, JARID2, and AEBP2 can bind to PRC2 and redirect the repressive complex to specific targets. DNMT-mediated methylation of DNA complements gene silencing. Non-canonically, EZH2 can methylate non-histone proteins leading to either gene silencing or activation and protein stability regulation. The non-canonical activity of EZH2 can be also methyltransferase independent and result from EZH2 direct interactions with several transcription regulatory proteins. See Figures 2–5 for details.
H3K27me3 is a marker of transcriptional repression, and EZH2-mediated epigenetic silencing of cancer suppressors contributes to the development of various malignancies, including hepatocellular carcinoma (HCC), 9 prostate cancer (PCa),10,11 breast cancer, 12 ovarian cancer, 13 lung cancer, 14 B-cell lymphomas, 15 glioblastoma, 16 colorectal cancer, 17 and melanoma. 18 Augmented expression of EZH2 in cancer often correlates with poor prognosis.19,20 Gain-of-function (GoF) mutations of the EZH2 SET domain, including Y646 (Y641 in mouse), have been associated with enhanced histone methyltransferase (HMT) activity and downregulation of cancer suppressor genes. 21 In hematological malignancies, changes in the EZH2 level are mainly caused by somatic mutations, either GoF mutations predominantly detected in lymphomas22,23 or loss-of-function (LoF) mutations mostly found in myeloid disorders.24,25 Analysis of LoF mutations in EZH2 suggests that EZH2 can act as a tumor suppressor in selected hematologic malignancies.24–26 Abnormal levels and activity of EZH2 in cancer cells can also result from various transcriptional and post-transcriptional modifications of EZH2, as well as cancer-type-specific alterations in signaling pathways (see below).
Several recent reviews summarize the canonical function of EZH2.8,27–29 In this review, we focus on the non-canonical role of EZH2 related to the methylation of non-histone proteins and methyltransferase-independent activity resulting from the direct binding of EZH2 to various nuclear and cytoplasmic proteins (Figure 1). These non-canonical roles of EZH2 are considered as a part of its oncogenic activity. We discuss the experimental approaches undertaken to inhibit EZH2 activity, and dual inhibitors and degraders of EZH2 protein are reviewed in more detail. We also summarize current data from clinical trials investigating EZH2 inhibitors as monotherapy or in combination with other agents for the treatment of various cancer types.
Non-canonical methyltransferase-dependent EZH2 activity
EZH2 activity contributes to gene repression through H3K27 methylation, and this activity is referred to as canonical. Methylation of various non-histone proteins is a part of a non-canonical EZH2 activity. It includes methylation of PRC2 components to enhance HMT activity and non-histone protein methylation participating in the positive and negative regulation of gene expression and the control of protein turnover.
Methylation of PRC2 components and cofactors to support canonical activity
Non-canonical methylation of the components of the PRC2 complex enhances its HMT activity (Figure 2(a)). Automethylation of EZH2 occurs in cis (intramolecularly) at three lysine residues K510, K514, and K515 that are close to each other on the conserved flexible loop containing many positively charged amino acids. 30 Utomethylation of EZH2 in response to cellular SAM concentration and the status of H3K27 methylation has been shown in vitro and in vivo as promoting HMT activity by increased accessibility of PRC2 to H3K27.30,31 In addition, EZH2 automethylation is supported by AEBP2, a PRC2 cofactor, and diminished by another PRC2 cofactor, JARID2. 31 EZH2 automethylation is important during stem cell reprogramming and differentiation, the processes that require de novo methylation of H3K27. 30 A triple mutation of EZH2 at K510, K514, and K515 preventing automethylation reduces the methylating activity of PRC2, specifically conversion of H3K27me2 to H3K27me3, which is a rate-limiting step.30,31

PRC2-dependent methylation of non-histone proteins. (a) The autoregulatory feedback loop, in which EZH2 methylates the components of PRC2 complex to enhance H3K27 trimethylation and repression of PRC2 target genes. (b) EZH2 methylates EloA, the RNA polymerase II transcription elongation factor, to attenuate transcription. Methylated EloA may either recruit its repressive reader (red circle) or evict a positive transcription factor (green circle). (c) PRC2-dependent gene repression can be enhanced by direct methylation of transcription factors such as GATA4.
Apart from automethylation, EZH2 can mediate SUZ12 methylation, which is diminished by AEBP2.30,31 Methylation of JARID2 at K116 is another PRC2-dependent non-canonical activity of EZH2. JARID2me3 by binding to an aromatic cage of EED promotes allosteric activation of PRC2 catalytic activity. 3
PRC2-dependent non-canonical activity of EZH2 in the repression of transcription
EZH2 in the PRC2 complex can repress transcription not only by trimethylation of H3K27 but also by methylation of non-histone targets (Figure 2, Table 1). Elongin A (EloA), a subunit of ternary complex EloA-EloB-EloC, which interacts with RNA polymerase II and stimulates transcription elongation, is monomethylated by EZH2 at K754. 32 This methylation complements PRC2-dependent gene silencing and is important for normal embryonic stem cell differentiation. 32 In the proposed model, methylated EloA may either recruit its repressive reader (red circle) or evict a positive transcription factor (green circle), in both cases downregulating the expression of target genes (Figure 2(b)).
EZH2-mediated methylation of non-histone targets.

AR, androgen receptor; CRPC, castration-resistant prostate cancer; DLC1, deleted in liver cancer 1; EED, embryonic ectoderm development; EloA, elongin A; ESC, embryonic stem cell; EZH2, enhancer of zeste homolog 2; FOXA1, forkhead box A1; GATA4, GATA-binding protein 4; HMT, histone methyltransferase; JARID2, jumonji and AT-rich interaction domain containing 2; me1,2,3, mono-, di-, trimethylation; N/A, not available; NKT, natural killer T cell; NSCLC, non-small-cell lung cancer; PLZF, promyelocytic leukemia zinc finger; PRC2, Polycomb repressive complex 2; RORα, retinoic acid-related orphan nuclear receptor alpha; STAT3, signal transducer and activator of transcription 3; SUZ12, suppressor of zeste 12; TCF1, T-cell factor 1; TNBC, triple-negative breast cancer.
EZH2 as a part of PRC2 can also directly methylate transcription factors to diminish their activity in normal lineage-specific differentiation and during cancerogenesis (Figure 1, Table 1). It has been demonstrated that PRC2 can interact with GATA-binding protein 4 (GATA4), a transcription factor crucial for heart development. Methylation of GATA4 at K299 attenuates its acetylation by transcriptional coactivator p300, thus repressing the transcriptional potency of GATA433 (Figure 2(c)). An interaction of EZH2 with GATA4 has been also detected in cardiac rhabdomyosarcoma. 46
Non-canonical methyltransferase activity of EZH2 in gene activation
Growing evidence indicates that EZH2 can also act as a transcriptional co-activator (Figure 3, Table 1). This activity can be mediated by its trans-activating domain (TAD) that spans amino acids 135–254. 47 In normal conditions, TAD is structurally blocked but can be unlocked by cancer-specific phosphorylation of EZH2. 47 EZH2 can be phosphorylated at serine 21 (S21) by pAKT as shown in breast cancer, 48 castration-resistant prostate cancer (CRPC), 11 glioblastoma stem cell subpopulation, 34 and colorectal cancer. 38 EZH2 phosphorylation at S21 reduces the EZH2 affinity to H3K27 leading to de-repression of PRC2 target genes. 48 It has been shown in PCa cells that the chromatin loci enriched in phosphorylated EZH2 are devoid of SUZ12, EED, and other components of PRC2, but contain RNA polymerase II and di- and tri-methylated H3K4, which is a mark of transcriptionally active chromatin. 11 Phosphorylation of EZH2 at S21 turns EZH2 into a transcriptional co-activator promoting proliferation, invasion, and stem-like properties of cancer cells by methylating specific transcription factors, such as androgen receptor (AR), 11 signal transducer and activator of transcription 3 (STAT3),34,36 and β-catenin 38 (Figure 3, Table 1). It has been shown in colorectal cancer that S21-phosphorylated EZH2 trimethylates β-catenin at K49, which induces β-catenin interaction with RNA polymerase II and T-cell factor 1 and modulates the expression of genes involved in cell migration and metabolic processes. 38 While the pEZH2-STAT3 interaction is common for various cancer types, its molecular effects are context dependent. For instance, EZH2-mediated STAT3 monomethylation at K180 has been identified in the glioblastoma stem-like cell subpopulation characterized by highly active phosphoinositide 3 kinase (PI3K)/AKT pathway and increased level of S21-phosphorylated EZH2. 34 EZH2-mediated methylation of STAT3 at K180 in breast cancer cells reinforces its nuclear retention and expression of STAT3 targets such as matrix metalloproteinase 2 and cyclin D1. 35 STAT3 dimethylation at K49 in colon cancer cells favors the expression of interleukin 6 (IL-6)-dependent genes. 36 In murine melanoma cells, the GoF EZH2Y641F mutant cooperates with STAT3 to drive the upregulation of a subset of interferon-regulated genes. 37

Non-canonical EZH2 methyltransferase activity promoting gene expression. Phosphorylation of EZH2 at S21 by pAKT has been recognized as a molecular switch that turns EZH2 into a transcriptional activator. pS21-EZH2 can methylate AR, STAT3, and β-catenin, which leads to an activation of gene sets supporting the progression of various cancers.
Non-canonical EZH2 methyltransferase activity involved in protein stability
Another non-canonical function of EZH2 is its contribution to the regulation of protein stability, particularly in methylation-dependent ubiquitination. In this mechanism, EZH2 methylates the protein substrate at lysine, which is subsequently selectively recognized by a specific adaptor linking monomethylated substrate to E3 ubiquitin ligase complex composed of damage-specific DNA-binding protein 1 (DDB1) and cullin 4 (CUL4). Such a mechanism of protein degradation has been shown for retinoic acid-related orphan nuclear receptor α (RORα) with DDB1 and CUL4-associated factor 1 (DCAF1) as an adaptor. 39 Monomethylation of RORα at K38 by EZH2 leading to RORα ubiquitination and degradation has been found in breast cancer cell lines 39 (Figure 4(a), Table 1). An elevated level of EZH2 correlates with a reduced level of RORα in breast tumor specimens further suggesting that EZH2 may facilitate RORα degradation, thus contributing to the repression of RORα target genes. 39 Interaction of EZH2 with a ubiquitin ligase complex has been also detected in natural killer T (NKT) cells. 40 In these cells, EZH2 monomethylates promyelocytic leukemia zinc finger transcription factor at K430 promoting its ubiquitination and degradation 40 (Figure 4(a), Table 1). EZH2 playing a role in protein stability has been also found in liver cancer cells. 41 Deleted in liver cancer 1 (DLC1) can form a complex with EZH2 in the cytoplasm, which results in DLC1 methylation at K678 followed by the reduction of its steady-state level (Figure 4(a), Table 1).
Ubiquitination is balanced by deubiquitylating enzymes that play distinct roles in tumorigenesis. 49 It has been reported that EZH2-mediated methylation of forkhead box A1 (FOXA1) at K295 is recognized by protein BUB3, which subsequently stimulates the recruitment of ubiquitin-specific peptidase 7 to induce deubiquitylation, thereby preventing FOXA1 degradation along the proteasome proteolytic pathway 42 (Figure 4(b), Table 1). EZH2-enhanced stabilization of transcription factor FOXA1 has been found to promote cell cycle progression and growth of PCa. 42 In HCC, EZH2 associated with a long noncoding RNA called lnc-β-Catm and β-catenin induces β-catenin methylation at K49, which, in turn, suppresses the ubiquitination of β-catenin sustaining a high activity of the WNT/β-catenin pathway. 50

EZH2 methyltransferase activity is involved in protein turnover. (a) EZH2 mediates monomethylation of RORα, PLZF, and DLC1. Methylated proteins are then recognized by DCAF1 associated with the CUL3/CUL4 E3 ligase complex and directed to proteasomal degradation. (b) EZH2-mediated methylation of transcription factors such as FOXA1 stimulates their deubiquitylation and prevents their degradation, which in the case of FOXA1 contributes to prostate cancer development. (c) EZH2-p38α cooperation is needed to enhance the activity of the PI3K/AKT pathway in breast cancer. p38α phosphorylates EZH2 to trigger its cytoplasmic localization, which, in turn, causes methylation of p38α, thereby enhancing its activity and stability.
Other non-canonical EZH2 methyltransferase activities in cytoplasmic localization
The enhanced activity of the PI3K/AKT pathway has been reported to be associated with increased EZH2 levels in triple-negative breast cancer (TNBC) progression. 43 The molecular mechanisms are complex and involve EZH2-p38α cooperation in the AKT activation (Figure 4(c), Table 1). EZH2 phosphorylation at T367 by p38α in the nucleus is needed for the cytoplasmic localization of EZH2. Cytoplasmic pEZH2, in turn, induces p38α methylation at K165 and trimethylation at K139 enhancing p38α stability and activity leading to AKT activation. Thus, the study suggests that EZH2 exerts a dual function in promoting breast cancer through the H3K27me3 silencing mechanism and by methylation of p38α. 43
The methylation of cytoplasmic integrin adaptor talin1 is another example of the non-canonical EZH2 methyltransferase activity in cytoplasmic localization. 44 This extranuclear function of EZH2 affects the binding of talin1 to F-actin, which disturbs the migration of neutrophils and dendritic cells, and immune responses. Further study has shown that this cytoplasmic activity of EZH2 can promote tumorigenesis through the interaction with the cytoskeleton remodeling effector, VAV. 45
Non-canonical methyltransferase-independent activity of EZH2
A growing number of methyltransferase-independent EZH2 activities have been reported recently. This chapter will focus only on the most prominent EZH2 interactions leading to stabilization of target proteins, and fine-tuning the activity of various oncogenic pathways. The interactions of EZH2 with non-coding RNAs that additionally modify the mode of action of EZH2 are beyond the scope of this review and are summarized elsewhere.4,5
Protein stabilization by EZH2 binding
EZH2 can control protein turnover either in a methyltransferase-dependent (Table 1, Figure 4) or non-catalytic manner (Table 2). EZH2 can bind directly to specific target proteins to impede their ubiquitination and proteasomal degradation.51,52
Methyltransferase-independent EZH2 interactions.
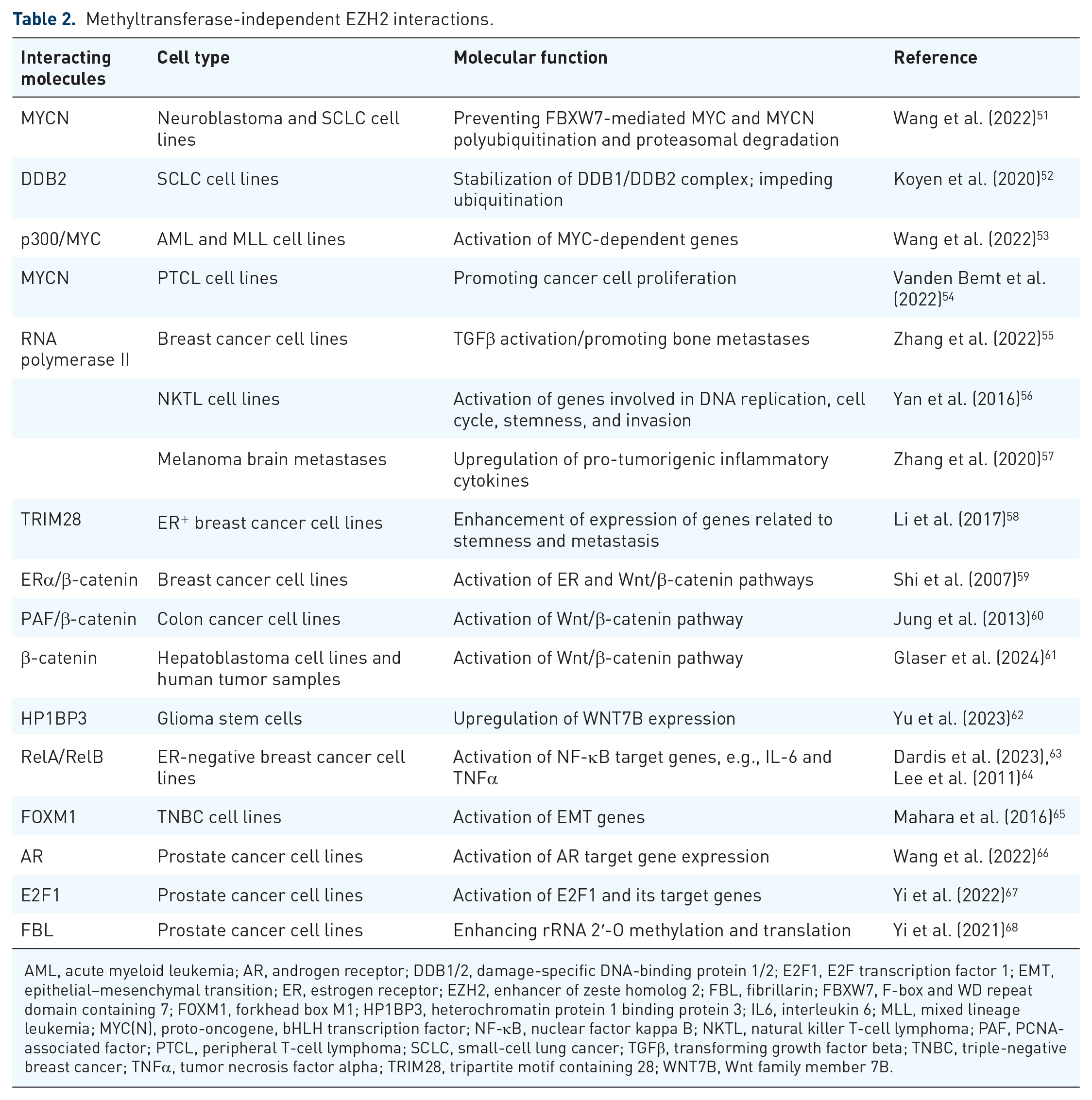
AML, acute myeloid leukemia; AR, androgen receptor; DDB1/2, damage-specific DNA-binding protein 1/2; E2F1, E2F transcription factor 1; EMT, epithelial–mesenchymal transition; ER, estrogen receptor; EZH2, enhancer of zeste homolog 2; FBL, fibrillarin; FBXW7, F-box and WD repeat domain containing 7; FOXM1, forkhead box M1; HP1BP3, heterochromatin protein 1 binding protein 3; IL6, interleukin 6; MLL, mixed lineage leukemia; MYC(N), proto-oncogene, bHLH transcription factor; NF-κB, nuclear factor kappa B; NKTL, natural killer T-cell lymphoma; PAF, PCNA-associated factor; PTCL, peripheral T-cell lymphoma; SCLC, small-cell lung cancer; TGFβ, transforming growth factor beta; TNBC, triple-negative breast cancer; TNFα, tumor necrosis factor alpha; TRIM28, tripartite motif containing 28; WNT7B, Wnt family member 7B.
In neuroblastoma and small-cell lung carcinoma (SCLC), EZH2 interacts with two oncogenic proteins, MYCN and MYC, respectively. EZH2 prevents F-box and WD repeat domain-containing 7 (FBXW7)-mediated MYCN polyubiquitination and proteasomal degradation, and EZH2 depletion decreases MYCN transcriptional activity. 51
Methyltransferase-independent interference of EZH2 with ubiquitin-proteasome pathway leads to increased stability of oncogenic proteins such as DDB2. DDB2 recognizes DNA lesions and recruits the nucleotide excision repair factors. 52 DDB2 is then ubiquitylated and targeted for degradation by the CUL4-RING E3 ubiquitin ligase complex. EZH2 localizes at DNA damage sites in SCLC cells and directly binds to DDB2. This interaction promotes the stabilization of the DDB1–DDB2 complex and enhances DNA repair, contributing to cisplatin resistance of SCLC. 52
EZH2 non-canonical role in the activation of gene expression
EZH2 methyltransferase-independent activity can be also executed after binding general transcriptional regulators or specific transcription factors (Table 2). These interactions are highly context dependent and mediated by distinct mechanisms. In these interactions, different domains of EZH2 can participate, including TAD,59,63,66 central region with C-X-C cysteine-rich domain, 60 and carboxyl end with pre-SET and SET domains (Figure 5(a)). 58

The non-canonical role of EZH2 in the co-activation of transcription. (a) Schematic representation of EZH2 functional domains. Gray ovals represent various kinases responsible for specific EZH2 phosphorylation. (b–e) To enhance gene expression, EZH2 can bind general transcriptional regulators or specific transcription factors. (b) Interaction of EZH2 with MYCN stimulated by CDK1-dependent EZH2 phosphorylation at T487 in PTCL leads to MYCN-dependent gene upregulation, including EZH2. (c) The binding of EZH2 to RNA polymerase II leads to overexpression of ITGB1, which enhances TGFβ signaling in breast cancer. JAK3-mediated EZH2 phosphorylation at Y244 supports EZH2 binding to RNA polymerase II in NKTL, whereas SRC-dependent phosphorylation of EZH2 at Y696 contributes to c-JUN upregulation. (d) Context-dependent interplay between EZH2 and NF-κB. In ER-positive breast cancer, EZH2 represses NF-κB target genes through the canonical pathway, whereas in the absence of ER EZH2 stimulates NF-κB target genes such as IL-6 and TNF, which through a positive feedback loop causes constitutive activation of NF-κB signaling. (e) Interaction of EZH2 with E2F1 facilitates the self-activation of E2F1 expression, which, in turn, upregulates CDCA8 in prostate cancer.
EZH2 can interact with general transcriptional regulators and specific transcription factors to enhance gene expression (Figure 5(b)–(e)). It has been demonstrated that phosphorylated EZH2 with active TAD can directly interact with transcriptional co-activator p300 to stimulate gene expression. 47 Active TAD is used to recruit a co-activator to enhance MYC-dependent genes in acute myeloid leukemia (AML) and mixed lineage leukemia (MLL) cell lines. 53 EZH2 has been also identified as a transcriptional co-activator for MYCN-dependent gene expression in peripheral T-cell lymphoma, including expression of EZH2 itself 54 (Figure 5(b)). A direct interaction between EZH2 and MYCN is stimulated by cyclin-dependent kinase 1-dependent EZH2 phosphorylation at T487. 54
EZH2 has been identified to function as a binding partner of RNA polymerase II (Figure 5(c)) to upregulate transcription of selected genes in breast cancer 55 and natural killer/T-cell lymphoma (NKTL) cell lines, 56 and to trigger melanoma brain metastases. 57 In breast cancer cells, the EZH2-polymerase II cooperation enhances the expression of ITGB1 encoding integrin β1, which leads to activation of the focal adhesion kinase and the transforming growth factor beta pathway promoting bone metastases. 55 The interaction of EZH2 with polymerase II in NKTL cells, supported by Janus kinase 3-mediated EZH2 phosphorylation at tyrosine 244 (Y244), upregulates genes involved in DNA replication, cell cycle, stemness, and invasion. 56 EZH2 phosphorylated at tyrosine 696 (Y696) by SRC non-receptor tyrosine kinase binds RNA polymerase II and induces c-JUN expression that promotes the expression of protumorigenic inflammatory cytokines that recruit immunosuppressive neutrophils to support melanoma brain metastases. 57 A complex of EZH2 with the subunits of chromatin remodeler, tripartite motif containing 28, and SWI/SNF positively regulates a set of genes associated with carcinogenesis, metastasis, and stemness as shown in ER-positive breast cancer cell lines. 58
The activation of the Wnt pathway can be achieved by direct interaction of EZH2 with β-catenin and estrogen receptor α (ERα) in breast cancer cells. 59 PCNA-associated factor (PAF) forms a complex with EZH2 and β-catenin to support Wnt signaling in colon cancer cells. 60 EZH2 directly interacts with β-catenin contributing to hepatoblastoma pathogenesis. 61 Interaction of EZH2 with heterochromatin protein 1 binding protein 3 (HP1BP3) in glioma stem cells epigenetically activates Wnt/β-catenin signaling through upregulation of the Wnt family member 7B (WNT7B). 62
In cancer, EZH2 can modulate the NF-κB pathway in a context-dependent manner. 69 In ER-negative basal-like breast cancer cells, EZH2 plays a transactivation non-canonical function by forming the ternary complex with a dimer RelA/RelB to support the constitutive expression of NF-κB target genes such as tumor necrosis factor and IL-6 (Figure 5(d)). 64 In ER-positive luminal-like breast cancer cells, ER recruits PRC2 to NF-κB target gene promoters, and expression of RelB and NF-κB-dependent genes is repressed by canonical EZH2 activity. 64 Thus, mutually exclusive occupancy of NF-κB target gene promoters associated with either activating or repressive function of EZH2 might require a context-specific therapeutic strategy for targeting EZH2 in breast cancer. In addition, the migratory and stem-like properties of breast cancer cells can be enhanced by EZH2 interactions with forkhead box M1 (FOXM1) transcription factor, which is supported by high levels of hypoxia-inducible factor subunit alpha in TNBC cells. 65
The molecular context determining the function of EZH2 has been also found in PCa. 70 EZH2 can occupy and induce gene expression from the promoters marked with H3K27 acetylation (H3K27ac), in contrast to promoters bound by PRC2 and marked with H3K27me3. This indicates that the local chromatin structure can determine the EZH2 activity. 70 EZH2 can activate the transcription of AR, and inhibitors of EZH2 combined with AR antagonists exert a strong synergy in suppressing PCa in vitro and in vivo. 70 EZH2 with active TAD can bind AR but also its constitutively active splice variant AR-V7. 66 EZH2 can enhance transcription of E2F1 by recruiting E2F1 to its promoter. An elevated level of E2F1, in turn, upregulates the cell division cycle-associated 8, a subunit of the chromosomal passenger complex, which plays an important role in mitosis and positively correlates with poor clinical outcomes of PCa patients (Figure 5(e)). 67 In addition to the non-canonical activities of EZH2 in the co-activation of transcription, its role in the regulation of translation in PCa cells has been demonstrated as a result of the direct functional interaction between EZH2 and fibrillarin, a SAM-dependent methyltransferase involved in pre-rRNA processing. 68
EZH2 inhibitors and degraders
EZH2 is considered an attractive therapeutic target for pharmacological interventions in cancer. Based on their mechanisms of action, EZH2 inhibitors can be categorized as (1) inhibiting EZH2 methyltransferase activity, (2) disrupting the interaction between PRC2 components, and (3) causing EZH2 degradation.
Inhibitors of EZH2 methyltransferase activity
The EZH2 methyltransferase activity can be inhibited by S-adenosylhomocysteine (SAH) accumulation or by competing with the methyl donor SAM for the binding pocket. S-adenosylhomocysteine hydrolase inhibitors such as 3-deazaneplanocin A (DZNep), first investigated in breast and colon cancer cell lines, 71 belong to inhibitors of EZH2 methyltransferase activity that cause SAH accumulation, which, in turn, inhibits SAM-mediated methyl transfer. It is not specific for EZH2 as other methyltransferases are also dependent on SAM as a methyl donor. Interestingly, a combination of DZNep and AC1Q3QWB, a selective disruptor of HOTAIR-EZH2 interaction, has been found more efficient than either agent alone in killing tumor cells in breast cancer and glioblastoma patient-derived xenograft models. 72
A number of highly selective EZH2 inhibitors, including GSK126, 73 EPZ-6438 (tazemetostat), 74 CPI-1205 (lirametostat), 75 PF-06821497, 76 and SHR2554, 77 directly inhibiting methyltransferase activity of EZH2 by occupying its SAM-binding site yielded promising results in preclinical studies and some of them entered clinical trials for the treatment of diverse cancer types (Table 3). While numerous clinical trials have been initiated or completed, only tazemetostat 74 has been approved by the Food and Drug Administration (FDA) for cancer treatment, specifically for epithelioid sarcoma and follicular lymphoma.78,79 Tazemetostat competitively binds to the SET domain of EZH2 to prevent binding of SAM. The above-mentioned AC1Q3QWB, an agent targeting HOTAIR-EZH2 interaction, has been recently shown to increase the efficacy of tazemetostat in inhibiting proliferation and migration of endometrial cancer cells, 80 whereas in combination with GSK-LSD1 lysine-specific demethylase 1 inhibitor, AC1Q3QWB has shown the enhanced anticancer activity in glioblastoma patient-derived xenograft models. 81 Results of the study performed on CRPC cells, but also referred to public data on diverse types of solid tumors with wild-type EZH2, have revealed that expression of DNA damage repair (DDR) genes, especially those encoding elements of the base excision repair pathway, is significantly correlated with tumor sensitivity to EZH2 inhibitors. 82 Mechanistically, EZH2 inhibitors block methylation of FOXA1, and this suppression of the non-canonical transactivation function of EZH2 contributes to reduced expression of DDR genes. In this way, EZH2 inhibitors may enhance the sensitivity of solid tumors to genotoxic stress, such as chemotherapy or radiation. Another example of preferential sensitivity to EZH2 inhibition has been reported recently in the context of ARID1A LoF mutations in bladder, ovarian, and endometrial tumor models. 83 Tulmimetostat (CPI-0209), an orally available, clinical-stage inhibitor of EZH1 and EZH2, which induces selected changes in gene expression in addition to an extensive reduction in H3K27me3 levels, has been found efficacious alone or in combination with cisplatin. 83 Inhibition of EZH2 acting in a synthetic lethal manner in ARID1A-mutated tumors has been previously shown as causing downregulation of PI3K/AKT signaling in ovarian clear cell carcinoma. 84 Mutation rates in the gene encoding a chromatin remodeler ARID1A is one of the highest across several tumor types. Therefore, EZH2 inhibition might be a potential therapeutic strategy for solid tumors with ARID1A LoF mutations.
Clinical trials of inhibitors of EZH2 histone methyltransferase activity alone or in combination with other anticancer agents.

Source: Data obtained from ClinicalTrials.org site (as of June 14, 2024).
Clinical trials labeled as terminated, withdrawn, or with unknown status or no longer available have been ruled out.
AML, acute myeloid leukemia; AR, androgen receptor; BCL, B-cell lymphoma; CRPC, castration-resistant prostate cancer; DLBCL, diffuse large B-cell lymphoma; EC, endometrial cancer; EED, embryonic ectoderm development; ES, epithelioid sarcoma; EZH2, enhancer of zeste homolog 2; FL, follicular lymphoma; GoF, gain of function; HC, hepatocellular carcinoma; HDAC, histone deacetylase; HMT, enhanced histone methyltransferase; ICI, immune checkpoint inhibitor; MM, multiple myeloma; N/A, not available; (N)HL, (non-)Hodgkin lymphoma; NSCLC, non-small-cell lung carcinoma; OC, ovarian cancer; OCCC, ovarian clear cell carcinoma; PARP, poly-ADP ribose polymerase; SCLC, small cell lung cancer; TCL, T-cell lymphoma; TCM, T-cell mesothelioma; TGFβ, transforming growth factor beta; UC, urothelial carcinoma.
Comparative analysis of the impact of EZH2 inhibitors on the pattern of posttranslational histone modifications between EZH2 sensitive and insensitive solid tumor cell lines has revealed that while H3K27me3 levels were similarly reduced by EZH2 inhibitors, H3K27 acetylation (H3K27ac) levels were markedly increased only in the insensitive cells. 89 Mechanistically, the MLL1 forms a complex with p300/CBP to facilitate histone acetylation (H3K27ac) in response to EZH2 inhibitors, and H3K27ac-associated transcriptional output contributes to the resistance to EZH2 inhibitors in solid tumors. The efficacy of EZH2 inhibitors can be improved by H3K27ac co-inhibition but it leads to oncogenic reprogramming such as MAPK activation in some cancers. This, in turn, highlights the need for a combination therapy after the patient stratification based on tumor-intrinsic expression of MLL1. The authors suggest that the combination therapy might expand the potential of EZH2 inhibitors from hematological cancers to solid tumors. 89 These and several other reports indicate that the efficacy of EZH2 inhibitors in solid tumors is highly context dependent, and overexpression of EZH2 is only one of the determining factors.
Tazemetostat has been evaluated in clinical trials in combination with drugs already approved for cancer treatment (Table 3). For example, a combination of tazemetostat with dabrafenib (BRAFV600 inhibitor) and trametinib (MEK inhibitor) has been evaluated in a phase I/II study in BRAF-mutant melanoma patients who progressed on BRAF/MEK inhibitor therapy (NCT04557956). Tazemetostat in combination with talazoparib, a poly-ADP ribose polymerase (PARP) inhibitor, has been evaluated in metastatic CRPC (NCT04846478), and the use of this combination is considered as one of the potential synergies between different molecular pathways. 90
EZH2 plays a critical role not only in cancer cells but also in cells of the tumor microenvironment. 91 It has been demonstrated that the modulation of EZH2 expression reprograms intratumoral regulatory T cells 92 and improves the efficacy of anti-CTLA-4 therapy. 93 Therefore, the EZH2 inhibition is considered a promising modality to enhance current immunotherapies. 94 Tazemetostat and other inhibitors of EZH2 methyltransferase activity have been combined with various drugs modulating immune response such as pembrolizumab (e.g., NCT03854474, NCT05467748, and NCT06022757) or ipilimumab (NCT03525795) in different types of cancers (Table 3).
Dual-acting EZH2 inhibitors
Several research groups have applied the concept of synthetic lethality to expand the EZH2 inhibitory activity by adding other anticancer pharmacophores to existing EZH2 inhibitory templates, thus creating dual-acting inhibitors. 95 Dual HDAC and EZH2 inhibitors have displayed a capacity to decrease cancer cell viability at micromolar concentrations in several cancer cell lines. 96 ZLD-2 designed as an inhibitor of EZH2 and bromodomain-containing protein 4 (BRD4) has shown potent antiproliferative activity against cells of solid tumors, including bladder, breast, lung, and pancreatic tumors. 97 The dual-acting inhibitor that can diminish the EZH2 methylation activity and affect the AR level has been found effective against PCa cells, including those resistant to enzalutamide. 98 The dual inhibitor of PARP and EZH2 activities has shown higher inhibitory potential against TNBC cells than the combination of olaparib and tazemetostat. 99 One of the recently synthesized dual-target PARP/EZH2 inhibitors has demonstrated a capacity to induce excessive autophagy and inhibit TNBC growth. 100 The dual inhibitor of HMTs G9A and EZH2 has demonstrated the capacity to induce chromatin changes followed by the transcriptional activation of the immunostimulatory gene network modulating the immune microenvironment and extending the survival of mice with ovarian cancer. 101 Most recently, a novel dual inhibitor of EZH2 and heat shock protein 90 has exerted promising activity against temozolomide-resistant glioblastoma. 102
Agents disrupting PRC2 composition
Knowing the dependence of EZH2 activity on the scaffolding provided by EED, various inhibitors of the EZH2-EED interaction have been developed, recently reviewed in the paper of Bao et al. 103 Astemizole, a drug approved by the FDA, has been identified as an agent destabilizing the PRC2 complex in cancer cells by the disruption of the EZH2-EED interaction. 104 An agent EED226 can form with EED-EZH2, a ternary complex capable of inhibiting a PRC2 activity, and also PRC2 containing a mutant EZH2 protein resistant to SAM-competitive inhibitors. 105 MAK683, another selective inhibitor of EED, has been found active in cancer cells with EZH2-mutation-driven acquired resistance to EZH2 methyltransferase inhibitors. 106 Due to its balanced pharmacokinetics/pharmacodynamics profile and efficacy, it has been selected for clinical development and it is currently in phase I/II study for the treatment of patients with diffuse large B-cell lymphoma (DLBCL) and advanced solid tumors (NCT02900651).
EZH2 degraders
The studies showing the ineffectiveness of inhibitors of EZH2 catalytic activity in several cancer types but cell sensitivity to a complete depletion of EZH2 protein provide the rationale for a pharmacological degradation of EZH2 as a therapeutic strategy for treating EZH2-dependent cancers. Inhibitors that covalently bind cysteine residues in the SET domain of EZH2 can cause an inability to form disulfide bonds resulting in inappropriate folding that triggers EZH2 degradation by carboxyl terminus of Hsp70-interacting protein-mediated polyubiquitination. Two such compounds, gambogenic acid derivative GNA002 that binds C668 of EZH2 and IHMT-337 that binds C663, have shown specific cytotoxic effects in head and neck cancer cell lines and mouse xenografts, 107 and TNBC and DLBCL cell lines and xenografts, 108 respectively. Most recently, a novel EZH2 degrader with the capacity to form the covalent bonding with C663 in the SET domain of EZH2 and potent antiproliferation activity in B-cell lymphoma and TNBC cell lines has been reported. 109
Using targeted protein degradation (TPD) technology that has gradually become widespread and potentially beneficial in terms of therapy,110,111 specific EZH2 degraders have been developed to suppress all types of EZH2 activities in cancer. A first-in-class EZH2 selective degrader, hydrophobic tag-based MS1943, can induce TNBC cell death associated with the activation of the unfolded protein response pathway and prolonged endoplasmic reticulum stress. 112 Bifunctional degraders, also known as proteolysis-targeting chimeras (PROTACs), contain a specific ligand for a distinct E3 ubiquitin ligase, and another ligand specific for a target protein, and an optimized chemical linker connecting the two.113,114 Compared to small inhibitors of enzymatic activity that need to steadily occupy an active site of the enzyme, PROTACs by binding to a protein of interest trigger its irreversible degradation. Although approximately 600 different E3 ligases have been identified in human cells, only a few of them, including cereblon (CRBN), von Hippel-Lindau (VHL), and murine double minute 2, have been used to design PROTACs against oncogenes.115–118 VHL-recruiting compounds named YM181 and YM281 designed to degrade EZH2 have shown potent anticancer activity in various subtypes of lymphomas. 119 Other EZH2 degraders recruiting VHL, MS8815, and MS8847 have effectively inhibited the growth of TNBC and AML cells.120,121 Three other PROTACs such as E7, 122 U3i, 99 and MS17753 have been designed to cooperate with CRBN E3 ligase. U3i exerts a high specificity toward EZH2 degradation in TNBC cells while sparing normal cells. 99 E7 and MS177 have shown wider specificity. E7 is also capable of degrading EED and SUZ12 in various cancer cell lines. 122 MS177, in turn, can effectively deplete both canonical EZH2-PRC2 and noncanonical EZH2-cMYC complexes leading to suppression of cancer cell proliferation. 53 In multiple myeloma cells, MS177 has shown the capacity to reactivate PRC2-repressed genes and genes associated with the immune response with the simultaneous suppression of c-MYC-associated oncogenes. 123
The above reports suggest that EZH2 can be targeted for degradation using different approaches (Figure 6). Several EZH2 degraders, their mode of action, and tumor specificity, are summarized in Table 4.

Mechanism of action of EZH2 degraders. (a) Agents that bind the sulfhydryl groups in the SET domain of EZH2 trigger (CHIP)-mediated ubiquitination and degradation of EZH2. (b) PROTAC which consists of ligand-binding EZH2 and ligand for E3 ligase, both connected with a linker, mediates polyubiquitylation and proteasomal degradation of EZH2. (c) Hydrophobic tagging of EZH2 triggers destabilization of the protein followed by degradation. After each cycle of reaction, EZH2 degraders are being regenerated.
EZH2 degraders investigated in various cancers.

AML, acute myeloid leukemia; CHIP, co-chaperone C terminus of Hsp70-interacting protein; CRBN, cereblon; DLBCL, diffuse large B-cell lymphoma; EZH2, enhancer of zeste homolog 2; NSCLC, non-small-cell lung cancer; TNBC, triple-negative breast cancer; VHL, von Hippel-Lindau.
The challenges of targeting EZH2 in cancer
Numerous small-molecule agents that inhibit EZH2 methyltransferase activity or block the formation of active PRC2 complex have been examined in various types of cancer in preclinical settings and clinical trials. Unfortunately, the inhibition of histone methylating activity of EZH2 fails to diminish the progression of several types of cancer. The target residence time of EZH2 inhibitors might be one of the factors influencing their cellular efficacy. 124 In addition, the emergence of resistant clones with acquired mutations at the interface of PRC2-inhibitor or in crucial epigenome factors with cooperative function in gene silencing125–128 is an inevitable limitation of durable clinical response. A detailed analysis of resistance mechanisms developed in the patients with adult T-cell leukemia/lymphoma enrolled for the valemetostat clinical trials has been published most recently. 129
As EZH2 inhibitors that only inhibit the enzymatic activity of EZH2 may fail to suppress its non-canonical methylation-independent functions, selective EZH2 degraders are being developed. Several drawbacks and challenges must be addressed to increase the effectiveness of TPD technology. Some cancer subtypes might be insensitive to EZH2 degradation. For instance, while MDA-MB-231 breast cancer cell line is resistant to EZH2 degradation, BT549 and MDA-MB-468 cell lines are sensitive. 112 The final effects of this treatment are determined by the basal EZH2 levels and cell dependencies on EZH2 activity. The context-dependent interactions between EZH2 and non-coding RNAs may also affect EZH2 activities.5,130 Other aspects that need to be considered include the limited degradation scope, low water solubility of degraders, and overall toxicity of EZH2 degrading agents. It also becomes clear that the identification of E3 ligases with higher cancer cell specificity is crucial for the effectiveness of TPD technology, and this issue is largely unexplored considering the large number of E3 ligases. Prolonged depletion of EZH2 can alter the epigenetic status leading to tumor progression as shown for glioblastoma. 131 As these effects have not been observed after the short-term depletion of EZH2, precise dosing regimens of anti-EZH2 agents in the clinic might be crucial for the effectiveness of treatment. Therefore, while TPD technology is recognized as a therapeutic approach with great clinical potential, several unresolved problems require further studies.
Conclusion
This review gathers recent knowledge regarding non-canonical multifaceted functions of EZH2 in cancer development and progression. EZH2-mediated PRC2-dependent and PRC2-independent methylation of non-histone proteins, and methyltransferase-independent interactions of EZH2 with various non-histone proteins leading to gene activation or repression, and influencing protein turnover, are highlighted. More research is necessary to identify non-canonical functionalities of EZH2 in a particular cancer type, and possible synthetic lethality between inhibition of EZH2 and other epigenomic abnormalities or cancer type-specific alterations in signaling pathways. It is also important to elucidate the still not well-recognized complexity of disturbances caused by the inhibition of aberrant EZH2 activity since EZH2 inhibition can also promote cancer. All this knowledge is crucial for the development of treatment strategies more specific for each malignancy. Considering the limited efficacy of epigenetic therapies and the risk of drug resistance, EZH2 degrading agents used alone or in combination with therapy targeting cancer-type specific protein(s) and/or immunotherapies have become more and more attractive.