
Abstract
SMARCA4 deficiency plays a critical role in the oncogenesis of various aggressive tumors that exhibit resistance to conventional chemotherapy and radiotherapy, posing significant challenges to clinical management. The pivotal role of SMARCA4 deficiency in tumorigenesis suggests the need for a paradigm shift from traditional tumor origin-based approaches to novel tumor-agnostic strategies focused on molecular alterations associated with SMARCA4 deficiency. This review explores potential targetable molecular changes and emerging therapeutic strategies for SMARCA4-deficient tumors. Molecular alterations related to SMARCA4 deficiency involve impaired genomic stability, defects in DNA mismatch repair, and elevated tumor mutation burdens, all of which suggest potential sensitivity to immune checkpoint inhibitors (ICIs). Recent studies indicate that combining ICIs with chemotherapy or anti-angiogenic agents as first-line treatments may offer clinical benefits for SMARCA4-deficient tumors. Furthermore, SMARCA4 deficiency epigenetically affects chromatin accessibility, alters the distribution of Polycomb group proteins on chromatin, and modulates histone acetylation, highlighting the potential efficacy of epigenetic regulators such as EZH2 and HDAC inhibitors. In addition, synthetic lethality strategies targeting vulnerabilities in SMARCA4-deficient tumors are promising therapeutic approaches, including inhibitors of SMARCA2, CDK4/6, ATR, CHK1, PARP, and the oxidative phosphorylation pathway. Based on current clinical evidence, ICI-based combination therapies represent the most promising first-line regimens for SMARCA4-deficient tumors. Although a theoretical basis supports the potential of tumor-agnostic therapy as a promising strategy for these tumors, several challenges remain in clinical practice. These include heterogeneous therapeutic responses across tumor types, safety concerns associated with synthetic lethality-based agents, and the absence of any histology-agnostic approved therapy for SMARCA4-deficient tumors. The continued development of novel therapeutics and further large-scale clinical evaluations are essential to overcoming these barriers.
Keywords
Introduction
SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A, member 4 (SMARCA4) is a critical ATPase subunit within the SWI/SNF complex, playing a crucial role in chromatin remodeling and gene expression regulation. 1 SMARCA4 deficiency was initially identified in patients with non-small-cell lung cancer (NSCLC) in 2003, where it was associated with reduced survival rates. 2 Since then, SMARCA4 deficiency has been observed across a diverse range of tumors, including central nervous system tumors, 3 sinonasal cancers,4,5 thoracic malignancies,6–9 gastrointestinal cancers,10,11 and multiple gynecological cancers.12–15
Currently, there are no standard guidelines for the diagnosis and treatment of SMARCA4-deficient tumors. Most clinical trials use SMARCA4 gene mutations detected by next-generation sequencing (NGS) or SMARCA4 protein deficiency identified by immunohistochemistry (IHC) as inclusion criteria. The 2021 WHO classification of lung tumors recommends a combined approach of morphological, immunohistochemical, and molecular testing for diagnosis. 16 These tumors often present as undifferentiated or poorly differentiated carcinoma, sarcomatoid, or rhabdoid-like morphology, with prominent nucleoli and mitotic activity.17–22 IHC is the most cost-effective and widely used method for evaluating tumors with suspicious morphology. SMARCA4 deficiency is identified by the absence of nuclear expression in tumor cells, with preserved expression in non-tumor tissue. If IHC confirms the loss of SMARCA4, sequencing is not necessary for diagnosis.16,23 NGS can validate reduced SMARCA4 expression and detect germline or somatic mutations, co-mutations, tumor mutational burden (TMB), microsatellite instability (MSI), and mismatch repair (MMR) status, supporting family risk assessment and treatment decisions. 24 SMARCA4 mutations are categorized into Class 1 mutations and Class 2 mutations. Class 1 mutations are associated with loss of protein function, including nonsense, truncating, frameshift, splice-site mutations, as well as homozygous deletions and fusions. Class 2 mutations include missense mutations and variants of uncertain significance. 8 The frequencies of Class 1 and Class 2 mutations in SMARCA4-NSCLC are both approximately 50%.8,25 There is a discrepancy in detection rates between IHC and NGS. In SMARCA4-mutated NSCLC cases, protein loss occurs in 81%–84% of Class 1 mutations but in only 7% of Class 2 mutations.7,8 Conversely, tumors with SMARCA4 protein loss may still carry wild-type SMARCA4, possibly due to non-mutational mechanisms such as epigenetic silencing. 26
Due to their aggressive nature and inherent resistance to conventional chemotherapy and radiotherapy, the treatment of SMARCA4-deficient tumors poses significant challenges, underscoring the urgent need for innovative therapeutic strategies. Given the widespread occurrence of SMARCA4 deficiency across various tumor types, tumor-agnostic therapy emerges as a promising strategy. Tumor-agnostic therapy represents a novel therapeutic paradigm that targets shared molecular alterations across multiple tumor types, as opposed to traditional oncology approaches, which are primarily based on tumor origin and histopathological features. A notable example of successful tumor-agnostic therapy is pembrolizumab, which has been approved for tumors characterized by high microsatellite instability (MSI-H), deficient mismatch repair (dMMR), or TMB ⩾10 mutations per megabase (Muts/Mb). Pembrolizumab has achieved an overall response rate (ORR) ranging from approximately 30% to 45%. Other examples include larotrectinib and entrectinib, which have demonstrated ORRs of 57%–79% in tumors harboring neurotrophin receptor kinase (NTRK) gene fusions. Dabrafenib in combination with trametinib has been used for tumors harboring B-raf proto-oncogene, serine/threonine kinase (BRAF) V600E mutations, achieving ORRs ranging from 31% to 69%. Selpercatinib, a tumor-agnostic agent targeting tumors with the Ret proto-oncogene (RET) gene fusions, has demonstrated ORRs of 44%–84%.27–29 These successful applications offer promising prospects for developing strategies to treat SMARCA4-deficient tumors. However, since SMARCA4 deficiency cannot be directly targeted, therapeutic approaches must focus on the molecular alterations resulting from or associated with SMARCA4 loss.
In this review, we conducted a literature search on PubMed to identify studies addressing SMARCA4 deficiency across tumor types, with a focus on its incidence, molecular alterations, biological mechanisms, synthetic lethal strategies, clinical trials, cohort studies, and case reports. To supplement the available evidence, we additionally searched Google Scholar to include relevant studies presented at academic conferences. Studies related to non-tumor conditions involving SMARCA4 deficiency or those investigating non-solid tumors were excluded from the analysis. By integrating preclinical and clinical evidence, this narrative review offers a comprehensive overview of potential therapeutic approaches and future research directions for SMARCA4-deficient tumors. In addition, we propose that a tumor-agnostic strategy may represent a promising approach for the treatment of SMARCA4-deficient tumors. We outline potential targetable molecular alterations in SMARCA4-deficient tumors from a tumor-agnostic perspective and summarize the preclinical and clinical advances of relevant drugs. Finally, we discuss the challenges associated with developing tumor-agnostic therapies for SMARCA4-deficient tumors.
SMARCA4 deficiency across multiple tumor types
SMARCA4 deficiency plays a critical role in the initiation and progression of various malignancies. Specific deletion of Smarca4 in mouse club cells results in reduced chromatin accessibility, decreased expression of activator protein-1 (AP-1) transcription factor (TF) family members (Fos, Jun), and lung lineage-specific TFs (Nkx, Cebp, Gata). This significantly accelerates malignant transformation. 30 AP-1 TFs mediate nucleosome remodeling and chromatin opening by cooperating with other lineage-specific TFs at enhancers, thereby facilitating the recruitment of the SWI/SNF complex. Dysregulation of these processes contributes to oncogenesis. 31 In normal human mammary epithelial cells, the absence of SMARCA4 triggers the upregulation of cluster of differentiation 44 (CD44), a marker commonly associated with tumor stem cells. This leads to abnormal cell differentiation, epithelial-mesenchymal transition, and enhanced colony formation ability. 32 Moreover, studies in small-cell carcinoma of the ovary, hypercalcemic type (SCCOHT),13,14,33 thoracic undifferentiated tumors,23,34 and sinonasal teratocarcinosarcoma have identified SMARCA4 deficiency as a driver genetic event. 5
SMARCA4 deficiency occurs in various tumor types. Nearly 100% of SCCOHT tumors and all SMARCA4-deficient thoracic undifferentiated tumors (SMARCA4-UTs) exhibit SMARCA4 deficiency.6,12–14,16 It is also frequent in sinonasal teratocarcinosarcoma (73%), 5 undifferentiated/dedifferentiated endometrial carcinoma (37%–50%), 15 and NSCLCs (3.7%–6.8%).7–9 In addition, SMARCA4 deficiency has been reported in gastrointestinal undifferentiated/rhabdomyoid carcinoma (15.4%), 11 endometrial stromal sarcoma (8%), 12 atypical teratoid/rhabdoid tumor (AT/RT, 4%), 3 sinonasal carcinoma (4%), 4 ovarian clear cell carcinoma (4%), 12 and gastric cancer (0.5%) 10 (Figure 1). The widespread oncogenic role of SMARCA4 deficiency across multiple tumor types provides a pivotal foundation for the development of tumor-agnostic therapeutics.

Prevalence of SMARCA4 deficiency across different tumor types. The widespread deficiency of SMARCA4 across diverse malignancies provides a theoretical basis for tumor-agnostic therapeutics
Potential tumor-agnostic approaches for SMARCA4-deficient tumors
Since SMARCA4 deficiency cannot be directly targeted, therapeutic strategies should focus on molecular alterations that arise from or are associated with its absence. These include changes in genomic stability, DNA mismatch-repair status, TMB levels, and the immune landscape of the tumor microenvironment (TME), all of which are recognized as biomarkers for the efficacy of immune checkpoint inhibitors (ICIs). In addition, SMARCA4 deficiency has been shown to epigenetically modulate gene expression by affecting chromatin accessibility, providing a rationale for treatment with epigenetic modulators. Another common alteration in these tumors is the loss of SMARCA4 protein, which supports synthetic lethality strategies (Figure 2). Here, we summarize existing preclinical and clinical evidence on potential tumor-agnostic therapies for SMARCA4-deficient tumors.

Overview of molecular changes associated with SMARCA4 deficiency and corresponding treatment strategies for SMARCA4-deficient tumors. In the SWI/SNF complex, the intact SMARCA4 protein (highlighted in yellow) enhances chromatin accessibility through nucleosome remodeling. SMARCA4 deficiency disrupts the regulation of chromatin accessibility. Since SMARCA4 deficiency cannot be directly targeted, therapeutic strategies need to focus on secondary molecular alterations associated with SMARCA4 deficiency. These alterations mainly involve three aspects: (a) Genomic instability, characterized by elevated TMB and MSI levels, which serve as biomarkers for effective treatment with ICIs (red marker 1). (b) Epigenetic dysregulation, characterized by decreased chromatin accessibility and reduced histone acetylation level, which can be targeted with EZH2 inhibitors and HDAC inhibitors (red markers 2 and 3). (c) Lethal vulnerabilities from impairment of specific proteins or pathways, including SMARCA2, DDR pathway, CDK4/6, and OXPHOS pathway, which form the basis for synthetic lethality strategies and can be treated with corresponding inhibitors (red markers 4, 5, 6, and 7).
ICI-based therapies
Nargund et al. demonstrated that SMARCA4 specifically mediates the recruitment of MutS homolog 2 (MSH2) and MutS homolog 6 (MSH6) to super-enhancers regulating cell adhesion gene expression. These findings suggest that SMARCA4 plays a critical role in guiding MMR proteins to specific genomic regions. 35 A study involving 4508 colorectal cancer patients found a significant correlation between SMARCA4 deficiency and tumor dMMR status, 36 which is closely associated with MSI-H status.37–39 Similar results were observed in gastric cancer, where SMARCA4 mutations and decreased SMARCA4 expression were also related to MSI-H status.10,40
SMARCA4-deficient tumors typically exhibit high TMB (TMB-H). Dagogo-Jack et al. reported a median TMB of 12.2 Muts/Mb in 1169 patients with SMARCA4-deficient non-small-cell lung cancer (SMARCA4-NSCLC). 7 Another study involving 20 patients with SMARCA4-UT and 67 patients with SMARCA4-NSCLC reported a higher median TMB in SMARCA4-deficient tumors compared to SMARCA4-intact thoracic tumors (15.36 Muts/Mb vs 7.68 Muts/Mb). 26 Other research on 44,646 colorectal cancer patients also confirmed a correlation between SMARCA4 mutations and TMB-H. 41
Unlike other biomarkers associated with ICI efficacy, the expression of programmed cell death ligand 1 (PD-L1) in SMARCA4-deficient tumors is heterogeneous. In SMARCA4-NSCLC, the tumor proportion score (TPS) of PD-L1 ranges from 46% to 51%, with 15% to 24% of patients exhibiting high PD-L1 expression (TPS ⩾ 50%).7,42 In SCCOHT, approximately 90% of both tumor and stromal cells express PD-L1. 43 By contrast, SMARCA4-UT tends to show low PD-L1 expression, although this does not appear to completely abrogate ICI efficacy. For instance, Zhou et al. reported objective responses in three out of four PD-L1-negative SMARCA4-UT patients after receiving ICI-based combination therapy. 44 Another study reported that three SMARCA4-UT patients and one SMARCA4-NSCLC patient with PD-L1 expression ranging from 0% to 100% achieved partial responses (PR) after ICI monotherapy. 45 These findings suggest that PD-L1 expression may not be determinant for ICI response in SMARCA4-deficient tumors.
The TME of SMARCA4-deficient tumors varies by histologic subtypes. SCCOHT exhibits substantial infiltration of T cells, CD68+ macrophages, and tumor-infiltrating lymphocytes, suggesting an immunogenic landscape that may respond well to ICI treatment. 43 Conversely, SMARCA4-UT displays an “immune-desert” phenotype, characterized by low infiltration of CD3+ T cells, CD8+ T cells, and CD20+ B cells, and by the lack of tertiary lymphoid structures. 46
Current clinical evidence on immunotherapy for SMARCA4-deficient tumors is mainly concentrated in SCCOHT, NSCLC, thoracic undifferentiated tumors, gastric cancer, and pancreatic cancer (Table 1). A case report involving four patients with SCCOHT described complete response (CR) or PR to ICI monotherapy in all cases, with one patient showing a sustained PR for 6 months, and the other three remaining tumor-free for more than 18 months. 43 However, in other tumor types, objective responses to ICI monotherapy do not result in progression-free survival (PFS) or overall survival (OS) benefits. Zhou et al. found that despite SMARCA4-truncated NSCLC patients exhibiting better ORR and disease control rate (DCR) than SMARCA4 wild-type patients when treated with ICI monotherapy (ORR: 53% vs 20%, DCR: 56% vs 32%), the median PFS (mPFS) (6.6 months vs 4.4 months) and median OS (mOS) (13.0 months vs 12.0 months) in SMARCA4-mutant cases were not significantly prolonged. 9 Shinno et al. reported four PRs among 10 SMARCA4-deficient thoracic tumor patients who received ICI monotherapy. The ORR of first-line ICI monotherapy was better than that of later lines (80% vs 0%). Despite the encouraging initial responses, the mPFS for all patients was only around 2.4 months, suggesting a temporary benefit of ICI monotherapy. 45 Similar findings were also reported in other studies. In a non-randomized phase II basket trial evaluating pembrolizumab monotherapy in rare sarcomas, the ORR of 12 patients with SMARCA4-deficient sarcomas or malignant rhabdoid tumors was 25%, ranking the second highest among all tumors evaluated. However, the mPFS and mOS were only 2.0 and 2.8 months, with the lowest 6-month OS rate (33%) among all tumors assessed. 47
The clinical applications of ICIs in SMARCA4-deficient tumors.

ABCP, combination of atezolizumab, bevacizumab, carboplatin, and paclitaxel; ICI, immune checkpoint inhibitor; mOS, median overall survival; mPFS, median progression-free survival; NE, not evaluated; ORR, objective response rate; PD-1, programmed cell death 1; PD-L1, programmed cell death ligand 1; SCCOHT, small-cell carcinoma of the ovary, hypercalcemic type; SMARCA4-NSCLC, SMARCA4-deficient non-small cell lung cancer; SMARCA4-UT, SMARCA4-deficient thoracic undifferentiated tumor.
Although the benefit of ICI monotherapy is limited, ICI-based combination therapies hold promise for improving clinical outcomes (Table 1). Chemo-immunotherapy demonstrated good responses in SMARCA4-deficient thoracic tumors, with the mPFS of 7.5 months, more than double the 3.5 months in the chemotherapy alone cohort. Furthermore, paclitaxel-based chemo-immunotherapy exhibited a higher ORR and a longer mPFS compared to pemetrexed-based regimens (ORR: 48.2% vs 18.2%, mPFS: 10.0 months vs 7.3 months). 26 Similar findings were reported in a study of 46 SMARCA4-NSCLC patients, in which chemo-immunotherapy increased mPFS to 9.3 months, whereas mPFS for chemotherapy alone was 6.1 months. 48 A retrospective study involving 308 gastric cancer patients treated with ICI plus chemotherapy found that SMARCA4 mutations correlated with PR. 49 These results indicate a promising role of chemo-immunotherapy, which warrants further confirmation and research. In another study of 95 patients with Epstein-Barr virus (EBV)-associated gastric cancer undergoing immunotherapy, all four patients with SMARCA4 mutations achieved PR, including three who received anti-programmed cell death 1 (PD-1)/PD-L1 plus anti-cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) therapy and one who received anti-PD-1/PD-L1 monotherapy. 50 Among pancreatic cancer patients harboring SWI/SNF-related gene alterations, three of nine individuals receiving ICI had SMARCA4 mutations, and all exhibited objective responses. Two patients received anti-PD-1/PD-L1 monotherapy, with PFS of 15 months and over 33 months, respectively. The other patient received PD-L1 inhibitor-based combination therapy, achieving a PFS of over 36 months. By contrast, six patients with SWI/SNF-related alterations who did not receive ICI treatment showed no objective responses. 51 Integrating ICI with anti-angiogenic agents presents another promising approach. One SCCOHT patient maintained PR for more than 25 months after receiving camrelizumab and apatinib. 52 Three SMARCA4-UT patients achieved PR after treatment with the ABCP regimen (atezolizumab, bevacizumab, carboplatin, and paclitaxel), with the longest PFS exceeding 17 months (Table 1). 53 However, it is worth noting that while ICI-based combinations have shown promising outcomes and survival benefits in some tumor subtypes, their efficacy may vary across tumors owing to co-mutations, the TME, and the epigenetic landscape. Additional validation of ICI combination therapies is required through larger retrospective or prospective studies. Immune-related adverse events may also vary depending on the tissue of origin. Moreover, predictive biomarkers for response in SMARCA4-deficient tumors may extend beyond traditional ICI markers. Therefore, further development of biomarkers is necessary.
Several trials investigating ICI-based combination therapies are underway. The combined applications of anti-PD-1/PD-L1 antibodies with anti-CTLA-4 or anti-T-cell immunoreceptor with Ig and ITIM domain (TIGIT) antibodies, as well as the combination of PD-1/PD-L1 inhibitors with chemotherapy, anti-angiogenic agents, or other emerging therapeutics, represent a major focus of immunotherapy research in SMARCA4-deficient tumors (Table 2). Dual ICI regimens, such as nivolumab plus ipilimumab (NCT04416568) and atezolizumab plus tiragolumab (NCT05286801), are being tested in solid tumors with SMARCA4 deficiency. In addition, the PAVEP chemotherapy regimen (cisplatin, doxorubicin, etoposide, and cyclophosphamide) in combination with pembrolizumab (NCT04602377) is being evaluated in SCCOHT patients. Cemiplimab plus gemcitabine (NCT06790602) and a four-drug ABCP regimen (NCT07093762) are being evaluated in pancreatic adenocarcinoma with SWI/SNF alterations and SMARCA4-mutated NSCLC, respectively.
Clinical trials of potential tumor-agnostic therapies for SMARCA4-deficient tumors.

ABCP, combination of atezolizumab, bevacizumab, carboplatin, and paclitaxel; AE, adverse event; AT/RT, atypical teratoid/rhabdoid tumor; DCR, disease control rate; DLT, dose-limiting toxicity; DOR, duration of response; mOS, median overall survival; mPFS, median progression-free survival; MTD, maximal tolerated dose; ORR, overall response rate; PAVEP, combination of cisplatin, doxorubicin, etoposide, and cyclophosphamide; RDE, recommended dose for expansion; RP2D, recommended phase II dose.
Epigenetic-based therapies
EZH2 inhibitors
The SWI/SNF complex enhances chromatin accessibility, whereas the Polycomb group proteins counteract this activity by facilitating chromatin repression. 54 The Polycomb group proteins comprise two main complexes: Polycomb repressive complex 1 (PRC1) and Polycomb repressive complex 2 (PRC2). Enhancer of zeste homolog 2 (EZH2), the catalytic subunit of PRC2, induces chromatin condensation by mediating the trimethylation of histone H3K27.54,55
The delicate balance between Polycomb group proteins and the SWI/SNF complex is crucial for tumor suppression.56,57 SMARCA4 deficiency leads to the redistribution of Polycomb group proteins on chromatin, resulting in chromatin decompaction and transcriptional derepression at previously Polycomb-bound regions, ultimately altering gene expression profiles. 1 Cancers harboring defects in the SWI/SNF complex may develop a dependency on the activity of Polycomb group proteins. 58 Inhibition of Polycomb group proteins, particularly EZH2, has emerged as a promising therapeutic approach for SMARCA4-deficient tumors.
Existing EZH2 inhibitors include tazemetostat (EPZ-6438), valemetostat, EPZ005687, EPZ011989, GSK126, GSK343, 3-deazaneplanocin A (DZNep), HH2853, etc. Tazemetostat is the most well-established EZH2 inhibitor and has received FDA approval for treating follicular lymphoma and advanced epithelioid sarcoma, a tumor associated with the loss of SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily B, member 1 (SMARCB1). Preclinical study has demonstrated the antitumor activity of tazemetostat in SCCOHT xenograft mouse model (Table 3). 59 Ballabio et al. found that low SMARCA4 expression or the T910M mutation promoted the development of cerebellar medulloblastoma in mice. Treatment with DZNep inhibited the growth of mouse medulloblastoma cells and induced cell death in tumor spheroids within medulloblastoma organoids. Similar results were observed with tazemetostat and GSK-126 treatments (Table 3). 60 Dose-dependent inhibition of colony formation was also observed with tazemetostat or GSK-126 in other SMARCA4-mutated tumor cells, including ovarian, gastric, lung, and adrenal cortical cancer cells (Table 3).58,61 Furthermore, the loss of the SWI/SNF complex has been shown to suppress chemotherapy-induced double-strand DNA breaks by decreasing the expression and activity of topoisomerase IIα, contributing to chemotherapy resistance. 62 GSK126 and DZNep have shown promise in inducing cell cycle arrest, promoting apoptosis, and enhancing the sensitivity of SMARCA4-NSCLC cells to the topoisomerase II inhibitor etoposide (Table 3). 63
Preclinical research on novel therapeutic approaches.
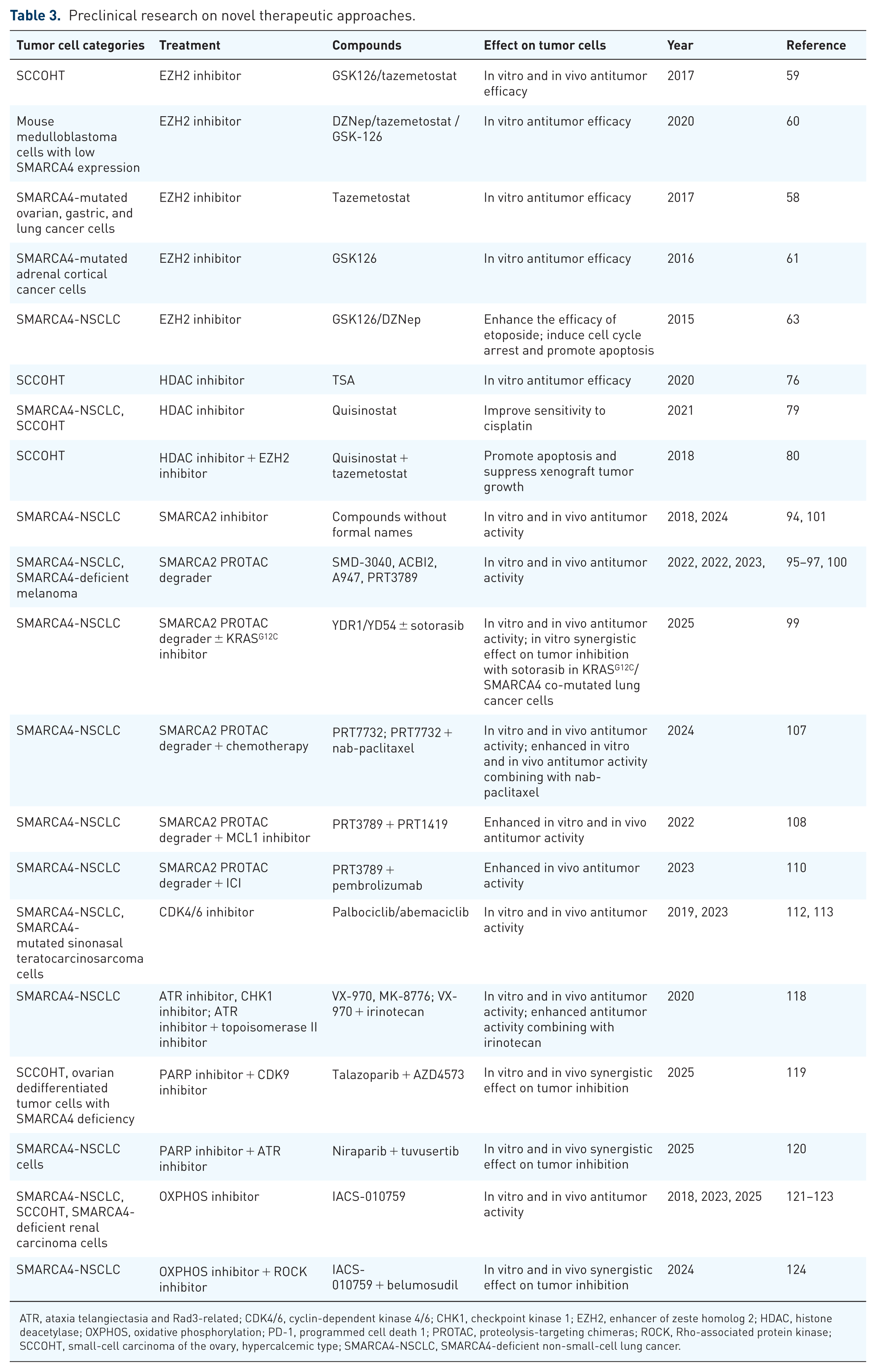
ATR, ataxia telangiectasia and Rad3-related; CDK4/6, cyclin-dependent kinase 4/6; CHK1, checkpoint kinase 1; EZH2, enhancer of zeste homolog 2; HDAC, histone deacetylase; OXPHOS, oxidative phosphorylation; PD-1, programmed cell death 1; PROTAC, proteolysis-targeting chimeras; ROCK, Rho-associated protein kinase; SCCOHT, small-cell carcinoma of the ovary, hypercalcemic type; SMARCA4-NSCLC, SMARCA4-deficient non-small-cell lung cancer.
A completed phase II study (NCT02601950) evaluated the efficacy of tazemetostat in patients with malignant rhabdoid tumors, including SCCOHT and thoracic sarcoma with SMARCA4 deficiency. Two of the 31 patients achieved PR, including one SCCOHT patient with a duration of response (DOR) of 32 weeks and one thoracic sarcoma patient with a DOR greater than 8 weeks. The study passed the stage 1 futility assessment, which required at least one PR or CR. However, it did not pass the stage 2 futility evaluation, which required confirmed PR or CR in at least five patients. 64 Another phase II clinical trial of tazemetostat in advanced solid tumors (NCT01897571) included two patients with SMARCA4-deficient malignant rhabdoid ovarian tumor and one patient with SMARCA4-deficient thoracic sarcoma, as well as patients with SMARCB1-deficient malignancies. One SMARCA4-deficient malignant rhabdoid ovarian tumor patient achieved PR, while another showed prolonged SD. In addition, one SMARCB1-negative malignant rhabdoid tumor patient achieved CR, and two SMARCB1-deficient epithelioid sarcoma patients showed SD. 65 The completed trial NCT03213665 investigated the efficacy of tazemetostat monotherapy in patients with mutant EZH2, SMARCB1, or SMARCA4. One patient with SMARCA4 deficiency, three patients with EZH2 mutation, and 16 patients with SMARCB1 deficiency were enrolled. A prolonged PR was observed in the SMARCA4-deficient non-Langerhans cell histiocytosis patient, with a PFS of 42.1 months. Four patients with SMARCB1 deficiency showed SD, with PFS ranging from 7.8 to 28.3 months. 66 In a phase II study of the EZH1/EZH2 dual inhibitor HH2853 for refractory solid tumors (NCT04390737), the cohort included one patient with high-grade pleural sarcoma harboring a canonical SMARCA4 splice-site mutation (c.2974-2A>T) and another patient with SMARCA4-UT. They received 400 and 600 mg of the drug, respectively. However, the outcome was unsatisfactory, as neither patient demonstrated a clinical response. 67 These studies suggest that despite tazemetostat showing activity in certain refractory tumors, there still exists heterogeneity across tumor subtypes to be further explored and overcome. To optimize the use of EZH2 inhibitors in these tumors, it is essential to identify the populations most likely to benefit and to elucidate the mechanisms underlying primary resistance.
Combining EZH2 inhibitors with other drugs may further enhance efficacy, such as in combination with ICIs. EZH2 inhibitors have shown the ability to modulate the tumor immune microenvironment. 68 EPZ005687 promotes the development and cytotoxic activity of natural killer (NK) cells. 69 GSK126 and GSK343 enhance NK cell-mediated elimination of hepatocellular carcinoma. 70 The combination of EPZ011989 and decitabine restores macrophage infiltration and reduces tumor burden in small-cell lung cancer mouse models. 71 In prostate cancer, EZH2 inhibition activates the double-stranded RNA-STING-interferon pathway, leading to upregulation of PD-L1 expression and increased infiltration of CD8+ T cells and M1 tumor-associated macrophages, thereby improving the effectiveness of cancer immunotherapy. 72 The combination of tazemetostat with nivolumab and ipilimumab (NCT05407441) is under evaluation (Table 2).
Histone deacetylase inhibitors
SMARCA4 deficiency has also been shown to affect histone acetylation. Histone acetylation facilitates chromatin opening and transcriptional activation. 73 The loss of SMARCA4 disrupts the recruitment of acetylated histones H3 and H4 to gene promoters, thereby suppressing gene expression.74,75 Histone deacetylase (HDAC) inhibitors promote gene transcription by increasing histone acetylation levels. SCCOHT and SMARCA4-deficient lung cancer cell lines are sensitive to the HDAC inhibitor trichostatin A (Table 3). 76 HDAC inhibitors tucidinostat and entinostat could potentiate antigen presentation and enhance antitumor efficacy of ICI mediated by CD8+ T cells.77,78 Quisinostat reversed chemotherapy resistance resulting from the downregulation of inositol 1,4,5-trisphosphate receptor type 3 (IP3R3) in SMARCA4-deficient tumor cells (Table 3). 79 Furthermore, Wang et al. found that SMARCA4 loss in SCCOHT precursor cells might make them reliant on HDAC activity to resist cell death signals and maintain a poorly differentiated state. Treatment with quisinostat induced apoptosis of SCCOHT cells, and the combination of quisinostat and tazemetostat synergistically increased histone H3K27 acetylation, suppressing tumor growth in SCCOHT mouse models (Table 3). 80
Currently, there is a lack of clinical reports on the efficacy of HDAC inhibitors in SMARCA4-deficient tumors. Based on clinical research in other tumors, HDAC inhibitor monotherapy shows limited benefit in solid tumors, while the combination of HDAC inhibitors with other therapies may improve efficacy.81–85
Other emerging therapeutic approaches
Although all these tumors exhibit SMARCA4 deficiency, directly targeting SMARCA4 deficiency is not feasible. In this case, synthetic lethality offers a promising alternative strategy. Synthetic lethality refers to a special interaction between two genes, where the loss of either gene alone does not lead to lethality, but the simultaneous deficiency of both genes results in cell death. 86 Exploiting synthetic lethal vulnerabilities in SMARCA4-deficient tumors has emerged as a crucial therapeutic strategy. These therapies primarily target SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A, member 2 (SMARCA2), cyclin-dependent kinases 4/6 (CDK4/6), DNA damage repair (DDR) pathways, and oxidative phosphorylation (OXPHOS).
SMARCA2 inhibitors and degraders
SMARCA2 functions as another catalytic subunit of the SWI/SNF complex and can partially compensate for SMARCA4 deficiency.74,87,88 SMARCA4-deficient tumor cells are highly dependent on SMARCA2 for survival.25,89 The synthetic lethal interaction between SMARCA4 and SMARCA2 underlies the susceptibility of SMARCA4-deficient tumors to SMARCA2 inhibition.90,91 Approaches targeting SMARCA2 include its inhibitors and proteolysis-targeting chimeras (PROTACs), which direct proteins to E3 ligases for proteasomal degradation.92,93 Preclinical studies have demonstrated that SMARCA2 inhibitors and PROTAC degraders induce cell death in SMARCA4-deficient/mutant melanoma and lung tumor cells (Table 3).94–97 Knockdown of SMARCA2 enhances the radiosensitivity of SMARCA4-NSCLC cells. 98 The SMARCA2 degraders YDR1 and YD54 have been reported to synergistically inhibit lung cancer cells with SMARCA4 and KRAS G12C co-mutations when combined with the KRAS G12C inhibitor sotorasib (Table 3). 99 In vitro and in vivo studies have shown that the SMARCA2 PROTAC degrader PRT3789 exerts significant anti-tumor activity in SMARCA4-NSCLC cells, while having no effect on the proliferation of SMARCA4 wild-type tumor cells (Table 3). 100 Currently, 11 SMARCA2 inhibitors and 5 SMARCA2 PROTAC degraders are under development and have been patented, highlighting the therapeutic potential of SMARCA2 inhibition. 101
A tumor-agnostic phase II study (NCT05639751) has been evaluating PRT3789 monotherapy and its combination with docetaxel in various advanced solid tumors with SMARCA4 deficiency (Table 2). Preliminary results of PRT3789 monotherapy from NCT05639751 were first presented at the ESMO conference in 2024. The study included 65 patients with NSCLC, pancreatic cancer, breast cancer, cholangiocarcinoma, colorectal cancer, esophageal cancer, and ovarian cancer. Thirty-four patients had SMARCA4 Class 1 mutations, 24 had Class 2 mutations, and 7 had SMARCA4 protein deficiency identified by IHC. Among the 46 efficacy-evaluable patients, 26 had NSCLC or esophageal cancer, and 7 of 26 showed tumor shrinkage. Two esophageal cancer patients achieved PR at doses of 24 and 120 mg, while one NSCLC patient achieved PR at 283 mg, and all of them had Class 1 mutations. Of the 26 NSCLC or esophageal cancer patients, 11 showed significant pharmacodynamic effects, four of whom achieved tumor shrinkage. No tumor responses were observed in the remaining 20 patients.102,103 Follow-up results for PRT3789 monotherapy were reported at the 2024 EORTC-NCI-AACR conference and the 2025 JSMO conference, with new patients of gastric cancer, small intestine cancer, and thoracic undifferentiated tumor included. At the AACR conference, four patients achieved PR. Two had esophageal cancer and received doses <283 mg, and another two had lung cancer and received doses ⩾283 mg. All four patients had Class 1 mutations. Among NSCLC and esophageal cases with Class 1 mutations, the most common co-mutant genes were TP53, CDKN2A, KEAP1, KRAS, and STK11. 104 At the JSMO conference, PRT3789 showed tumor responses and durable SD in NSCLC and upper gastrointestinal cancers. In addition to the four responders reported at the AACR conference, one gastric cancer patient with a Class 1 mutation who received a dose of ⩾283 mg also achieved PR. In NSCLC, the longest PFS was observed in a patient who received a dose of 48 mg and maintained SD for over 69 months. Among the three patients with esophageal or gastric cancer who achieved PR, one esophageal cancer patient receiving 24 mg had a PFS of over 9 months, another receiving 120 mg showed a PFS of approximately 30 months, and one gastric cancer patient receiving 500 mg had a PFS of over 12 months. 105 These results suggest that the efficacy of PRT3789 monotherapy across tumor types may be influenced by drug dose and pharmacodynamic effects. In addition, co-mutations, tissue-specific TME, SMARCA4 mutation type, and the extent of SMARCA4 loss may also modulate responses to SMARCA2 degraders.
PRT7732, a more potent and selective SMARCA2 degrader than PRT3789, has been shown to inhibit the growth of SMARCA4-NSCLC cells but not SMARCA4 wild-type or SMARCA2/SMARCA4 double-deficient cancer cells. Low-dose PRT7732 in combination with nab-paclitaxel has demonstrated robust tumor inhibition in mouse models (Table 3).106,107 A phase II clinical trial of PRT7732 (NCT06560645) in SMARCA4-deficient solid tumors is currently ongoing (Table 2).
Combining SMARCA2 degrader with a myeloid cell leukemia 1 (MCL1) inhibitor can increase cytotoxic activity (Table 3). 108 SMARCA2 degradation induces apoptosis, downregulates DNA replication and cell cycle pathways, and enhances antigen presentation, providing a theoretical foundation for combinations with CDK4/6 inhibitors, chemotherapy, and ICIs. In co-cultured peripheral blood mononuclear cells and SMARCA4-NSCLC cells, PRT3789 plus pembrolizumab promoted activation of CD8+ T cells, enhanced release of interferon gamma, interleukin-2, and granzyme B, and improved antitumor activity in mouse models compared with either monotherapy (Table 3).109,110 Based on the findings, a phase II clinical trial (NCT06682806) is exploring the efficacy of combining PRT3789 with pembrolizumab in patients with SMARCA4-mutant/-deficient solid tumors. The NCT05639751 study of PRT3789 combined with docetaxel remains under investigation. These indicate that SMARCA2 degraders will likely be used in combination with other therapies in SMARCA4-deficient tumors.
CDK4/6 inhibitors
Kinase-focused RNAi screen identifies a dependency of SCCOHT cells on CDK4/6 activation. 111 The repurposing of CDK4/6 inhibitors, such as palbociclib and abemaciclib, has shown antitumor activity in SMARCA4-NSCLC cells and SMARCA4-mutated sinonasal teratocarcinosarcoma cells (Table 3).112,113 Clinical application of abemaciclib plus nivolumab achieved sustained disease stabilization in an SCCOHT patient. 114 Another SCCOHT patient, whose tumor had progressed despite multiple prior treatments of surgery, chemotherapy, poly ADP-ribose polymerase (PARP) inhibition plus temozolomide, and anti-angiogenic therapy with immunotherapy, experienced tumor shrinkage after receiving combination therapy with an ICI, an anti-angiogenic agent, and a CDK4/6 inhibitor. The OS of the patient exceeded 5 years. 115
The ongoing phase II basket trial (NCT03297606) is investigating the efficacy of palbociclib monotherapy in SMARCA4-deficient solid tumors (Table 2). The tumor-agnostic Dutch DRUP and Australian MoST trials evaluated the efficacy of palbociclib or ribociclib monotherapy in patients with amplifications of CDK4, CDK6, CCND1, CCND2, CCND3, or complete loss of CDKN2A or SMARCA4. Across 139 enrolled patients, 11 had SMARCA4 inactivation. No objective responses were observed, and only 16 patients maintained SD. The mPFS for all patients was 4 months, and the mOS was 5 months, indicating limited clinical benefit of palbociclib or ribociclib monotherapy. 116 Therefore, it can be preliminarily speculated that in SMARCA4-deficient tumors, CDK4/6 inhibitors may be better suited for combination with other drugs, such as ICIs, chemotherapy, or anti-angiogenic agents.
DDR pathway inhibitors
SMARCA4 is indispensable for DDR. Replication protein A (RPA), the major single-stranded DNA (ssDNA)-binding protein, activates the ataxia telangiectasia and Rad3-related (ATR) pathway. RPA can also interact with the SWI/SNF complex. 117 The loss of SMARCA4 may disrupt the interaction between RPA and the SWI/SNF complex, intensify the RPA-ssDNA interaction, and thereby facilitate the activation of ATR and downstream checkpoint kinase 1 (CHK1), inducing an ATR dependency in SMARCA4-deficient tumors. SMARCA4-NSCLC cells exhibit vulnerability to ATR inhibitor (VX-970) and CHK1 inhibitor (MK-8776). Combining VX-970 with the topoisomerase II inhibitor irinotecan, which induces ssDNA breaks, results in improved tumor growth suppression (Table 3). 118
More recently, SMARCA4-deficient SCCOHT has been shown to have impaired homologous recombination repair, displaying a breast cancer susceptibility gene 1 (BRCA1)-deficient-like signature and sensitivity to PARP inhibitors. This is attributed to the disruption of RNA polymerase II (Pol II) elongation by SMARCA4 deficiency, resulting in the accumulation of R-loops and sequestration of BRCA1 to transcription complexes. The combination of PARP inhibitor talazoparib with CDK9 inhibitor AZD4573, which targets Pol II elongation, further suppresses cell proliferation in SCCOHT and ovarian dedifferentiated tumor cells, and enhances antitumor effects in an SCCOHT patient-derived xenograft (PDX) model (Table 3). 119 In SMARCA4-mutated NSCLC cells, the combination of PARP inhibitor niraparib with ATR inhibitor tuvusertib also exhibits a synergistic suppression of tumor growth (Table 3). 120
OXPHOS inhibitors
SMARCA4-NSCLC cells and SMARCA4-deficient renal carcinoma cells have been reported to be OXPHOS-dependent and sensitive to the OXPHOS inhibitor IACS-010759 (Table 3).121,122 Zhu et al. found that SMARCA4 deficiency decreased glucose transporter type 1 (GLUT1) expression in SCCOHT cells, resulting in glucose uptake impairment. As an alternative, the expression of glutamine transporter solute carrier family 38 member 2 (SLC38A2) was upregulated, augmenting glutamine uptake for OXPHOS. Inhibition of OXPHOS by IACS-010759 demonstrated suppressive effects in SCCOHT cells and PDX models. Supplementation with alanine, an amino acid that competitively binds to SLC38A2, showed a synergistic inhibitory effect with IACS-010759 (Table 3). 123 Recent preclinical study has suggested that combining the Rho-associated protein kinase (ROCK) inhibitor belumosudil (KD025) with IACS-010759 provides a novel strategy for achieving synergistic inhibition of SMARCA4-mutant tumors (Table 3). 124
However, phase I clinical trials of IACS-010759 (NCT03291938 and NCT02882321) in patients with advanced solid tumors and acute myeloid leukemia (AML) have demonstrated limited antitumor efficacy. Among 23 patients with solid tumors, there was one PR and eight cases of SD, while no responses were observed in 17 patients with AML. Common adverse events included elevated blood lactate, lactic acidosis, and peripheral neurotoxicity, which limited dose escalation and raised safety concerns. 125 Further research is needed to comprehensively evaluate the feasibility and safety of OXPHOS inhibitors.
Conclusion
Effective treatment of SMARCA4-deficient tumors represents an urgent clinical need; however, a consensus on optimal treatment strategies has yet to be established. This may be partly due to the fact that SMARCA4-deficient tumors have only gained significant attention in recent years. In addition, SMARCA4 deficiency itself cannot be directly targeted, which hinders the development of precision therapies. In this review, we propose that targeting molecular alterations associated with SMARCA4 deficiency and leveraging synthetic lethality strategies could offer new therapeutic avenues. Moreover, given the shared SMARCA4 deficiency in these tumors, treatment paradigms are expected to shift from traditional tumor origin- and histology-based approaches to candidate tumor-agnostic strategies.
SMARCA4 deficiency is associated with MSI-H, dMMR, and TMB-H. Existing reports suggest that ICI-based combination therapy may be a promising approach for SMARCA4-deficient tumors, and it seems to be more effective when used in first-line rather than in later lines. Ongoing clinical studies also indicate that ICI-based combination therapy is a key direction for therapeutic development in SMARCA4-deficient tumors, including the combinations of ICI with chemotherapy, anti-angiogenic drugs, epigenetic therapies, and integrated applications of different ICIs. TMB-H and dMMR/MSI-H are potential biomarkers for ICI efficacy in SMARCA4-deficient tumors. However, the correlation between ICI efficacy and PD-L1 expression in these tumors remains unclear. Li et al. suggested that patients with PD-L1 expression greater than 50% were likely to benefit from ICI monotherapy, while those with PD-L1 expression between 1% and 50% might be suitable for chemo-immunotherapy. For patients with PD-L1 less than 1%, first-line chemo-radiotherapy and second-line ICI monotherapy might be viable options. 68 However, their conclusions are primarily based on a small number of case reports and thus lack sufficient representation. After reviewing multiple studies, we observed no close correlation between PD-L1 expression and ICI efficacy in SMARCA4-deficient tumors. Large-scale studies are needed in the future to determine whether PD-L1 expression is necessary for ICI-based tumor-agnostic treatment in these tumors.
SMARCA4 deficiency also epigenetically changes chromatin accessibility and gene expression profile by altering EZH2 distribution and interfering with histone acetylation. EZH2 inhibitors exhibit antitumor activity against SMARCA4-deficient tumors both in preclinical and clinical research. HDAC inhibitors enhance the sensitivity of SMARCA4-NSCLC and SCCOHT cells to cisplatin and promote apoptosis of SCCOHT cells.
Targeting the synthetic lethality genes of SMARCA4 is theoretically applicable to all SMARCA4-deficient tumors. These targets mainly include SMARCA2, CDK4/6, ATR, CHK1, PARP, and the OXPHOS pathway, which show promising results in preclinical studies. Repurposing of these drugs is expected to expand treatment options for SMARCA4-deficient tumors and represents another frontier in drug development for these tumors.
There are several challenges in the treatment development process for SMARCA4-deficient tumors. First, despite all these tumors exhibiting SMARCA4 deficiency, whether it exerts an equally critical driver role in different tumor types requires further preclinical validation. Second, most therapies remain in the exploratory phase. Much of the clinical evidence originates from phase I/II trials, small-sample retrospective studies, or case reports. Some candidate compounds are still in the preclinical stage. Therefore, there is a lack of clinical evidence from large randomized controlled trials to support the approval of a tumor-agnostic approach for SMARCA4-deficient tumors. Moreover, the relative rarity of SMARCA4-deficient tumors limits patient enrollment in clinical trials and restricts the generalizability of the results. These factors constitute key limitations in advancing tumor-agnostic therapies for SMARCA4-deficient tumors. Multi-center collaboration may be a viable strategy to expand the patient cohort and improve the representativeness of findings. Third, safety concerns regarding synthetic lethality drugs need to be addressed, especially those targeting molecules with essential biological functions in normal cells, such as OXPHOS inhibitors. Finally, tumors with identical molecular alterations may exhibit different responses to the same drugs, which may be related to distinct co-occurring mutations, diversities in the TME, and variations in the epigenetic landscapes. For example, the ORRs of dabrafenib plus trametinib in patients with BRAF V600E-mutated melanoma, NSCLC, anaplastic thyroid carcinoma, biliary tract cancer, and high-grade glioma are 67%, 64%–68%, 69%, 47%, and 33%, respectively.126–130
Emerging evidence indicates that STK11 and KEAP1 are frequently co-deleted or co-mutated alongside SMARCA4 alterations. The frequency of concurrent deletions of SMARCA4, STK11, and KEAP1 in NSCLC is approximately 12%. In NSCLC cohorts characterized by co-mutations of SMARCA4 and STK11/KEAP1, patients demonstrated significantly worse PFS and OS under chemo-immunotherapy compared to those harboring either all wild-type alleles or SMARCA4 mutation alone.131–133 Notably, although ICI-based combination therapy is considered a promising strategy for SMARCA4-deficient tumors, patients with SMARCA4 deletions or mutations still exhibited inferior ORR and shorter PFS compared to those with wild-type SMARCA4.131,132
A bioinformatics-based pan-cancer analysis revealed that the infiltration of immune cells, such as CD4+ T cells, CD8+ T cells, B cells, neutrophils, dendritic cells, and macrophages, was negatively correlated with SMARCA4 expression, especially in glioblastoma multiforme, skin cutaneous melanoma, and sarcoma. Stromal cell infiltration was positively correlated with SMARCA4 expression in uveal melanoma, but negatively correlated in adrenocortical carcinoma, breast carcinoma, and colon adenocarcinoma. 134 In addition, a study of patients with SWI/SNF complex gene mutations, including SMARCA4, PBRM1, and ARID2, found that PD-1+ T-cell and CD38+ mature B-cell markers were upregulated in SMARCA4-low expressing colorectal and gastric cancers, but not in NSCLC or rectal cancer. 135 These findings suggest that despite shared SMARCA4 deficiency or low expression across these tumors, therapeutic responses, particularly to immunotherapy, may vary due to differences in the TME.
The epigenetic landscape of tumors has also been shown to influence sensitivity to various therapeutic agents.136–140 SMARCA4 expression was reported to correlate with DNA methyltransferase levels in thymoma, bladder urothelial carcinoma, and colon adenocarcinoma. 134 The expression of SWI/SNF-related genes was positively associated with RNA m6A regulators in several tumor types, including bladder urothelial carcinoma, hepatocellular carcinoma, and papillary renal cell carcinoma. 141 These factors could collectively influence the therapeutic response to the same drug across SMARCA4-deficient tumor subtypes. Despite the theoretical promise of tumor-agnostic treatment strategies, further detailed evaluation of their efficacy in various SMARCA4-deficient tumor types is warranted in the future.