
Abstract
Gastroesophageal adenocarcinoma (GEA) is a heterogeneous disease with a poor prognosis. Chemotherapy has been the cornerstone in treating metastatic diseases. Recently, the introduction of immunotherapy demonstrated improved survival outcomes in localized and metastatic diseases. Beyond immunotherapy, several attempts were made to improve patient survival by understanding the molecular mechanisms of GEA and several molecular classifications were published. In this narrative review, we will discuss emerging targets in GEA, including fibroblast growth factor receptor and Claudin 18.2, as well as the accompanying drugs. In addition, novel agents directed against well-known targets, such as HER2 and angiogenesis, will be discussed, as well as cellular therapies like CAR-T and SPEAR-T cells.
Introduction
Gastroesophageal adenocarcinomas (GEA) arise from the stomach, gastroesophageal junction and distal oesophagus. They are a significant worldwide health problem, ranking third on the tumour-related death list. 1 Especially in Western countries, the high mortality rate could be related to the tumour being often diagnosed as an advanced or metastatic disease 2 with a median overall survival (mOS) of 10–12 months when treated with systemic chemotherapy. Only 5.5% of patients diagnosed with metastatic GEA are alive at 5 years. 3 Additionally, even in the case of non-metastatic tumours, the rate of relapse after curative surgery is high, 4 leading to a poor outcome.
In metastatic tumours, systemic chemotherapy has been the standard of care for several years 5 ; however, recently, immunotherapy has been shown to be effective in GEA, improving the outcome in localized and metastatic diseases. 6 Several attempts have been made to improve patient survival by understanding the molecular mechanisms driving GEA and developing agents against these targets. In this regard, validated molecular classifications were introduced, which showed that GEA can no longer be considered a single entity but should be regarded as a heterogeneous disease with multiple subgroups. Among those classifications, the Cancer Genome Atlas (TCGA) 7 and the Asian Cancer Research Group (ACRG) 8 are the most important. Each classification distinguished four distinct gastric cancer (GC) subtypes with their own peculiarities and outcome. In particular, TCGA included Epstein-Barr virus positive (EBV; 9%), microsatellite instability (MSI; 21%), genomically stable (GS; 20%) and chromosomal instability (CIN; 50%), 7 whereas the ACRG considered MSI (23%), microsatellite stable with intact p53 (MSS/TP53; 36%), microsatellite stable with p53 mutations (MSS/TP53+; 26%) and microsatellite stable with epithelial-mesenchymal transition (MSS/EMT; 15%). 8
However, these classifications have yet to be used in everyday clinical practice. In this context, trastuzumab and ramucirumab have long been the only targeted agents approved and currently used in GEA, namely trastuzumab as first-line treatment for human epidermal growth factor receptor 2-(HER2)-positive GEA and ramucirumab in second-line treatment for all comers. 5 Recently, trastuzumab deruxtecan (T-Dxd) was approved for HER2-positive GEA in some countries. Nevertheless, several molecular pathways are under evaluation with the potential to become novel targets for drug development in GEA (Figure 1).
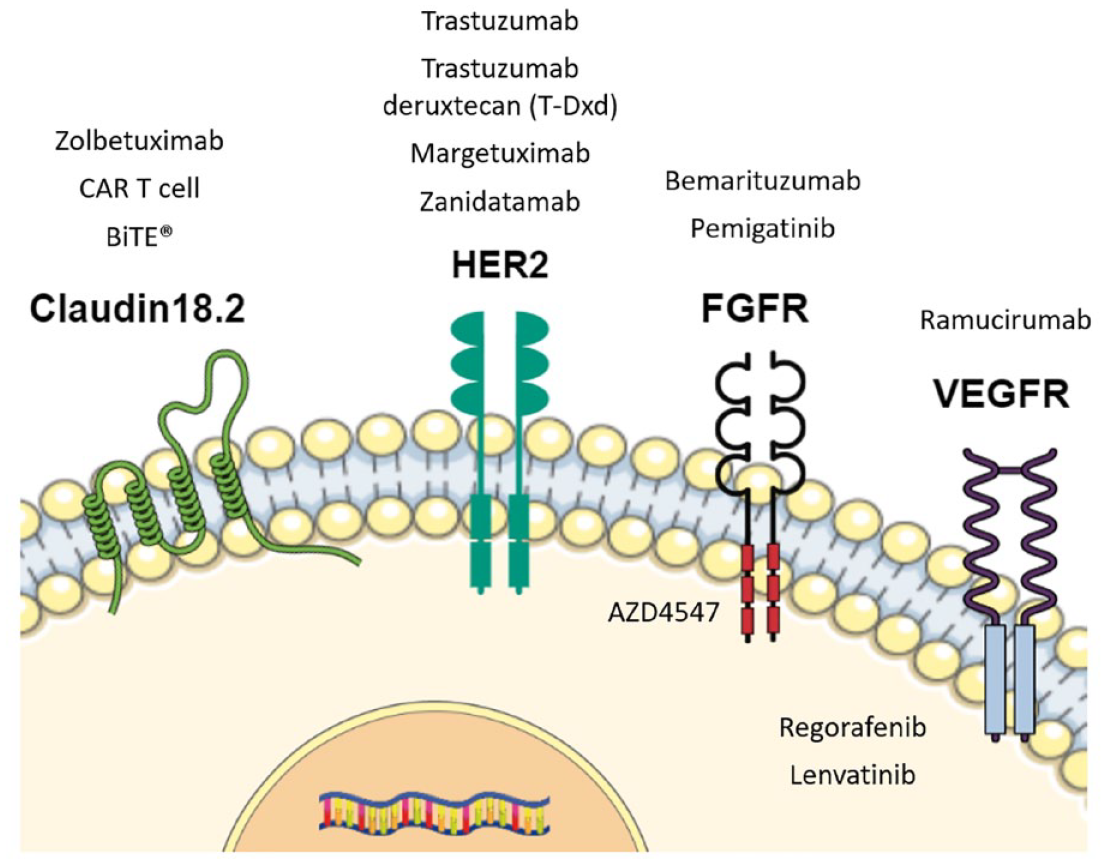
Main emerging molecular targets and therapeutic agents used and under evaluation in gastroesophageal adenocarcinoma.
This narrative review aims to depict the current status of emerging targets in GEA and state-of-the-art therapeutic developments, as well as future perspectives; it does not cover specific immune targets and the tumour microenvironment (TME). Thus, we report data about phase II and III trials in this field, showing phase I results only in case the first ones were unavailable.
Fibroblast growth factor receptor in GEA: Role and inhibitors
The fibroblast growth factor receptor (FGFR) family has been extensively studied as a potential target in GEA. 9 This family comprises four tyrosine kinase receptors (FGFR 1–4), which may form both homo or heterodimers, influencing angiogenesis, mitosis, differentiation, proliferation, changes in tissue homeostasis and cancer invasion processes via activation of several cellular pathways, including the RAS-RAF-MEK-ERK, PIK3CA-AKT-mTOR and JAK pathways. 9 Additionally, the activation of the FGFR pathway plays a role in endothelial cell proliferation and differentiation, both important for embryonic development, wound healing and intra-tumoral angiogenesis. Alterations in the FGFR pathway were reported in several tumours, including GEA. 10 Among all the possible alterations occurring in the FGFR pathway, FGFR1 mutations, FGFR2 amplifications and FGFR3 rearrangements are the most common in GEA (ranging 3–15%).11–13 These alterations lead to continuous activation of the FGFR pathways. In particular, FGFR2 amplification results in protein hyperexpression, which can massively interact with the ligands; chromosomal translocations alter the FGFR3 gene leading to rearrangements and aberrant proteins, which are constitutively active in the absence of ligand, like in the case of FGFR1 mutations too. 14 Then, concomitant alterations in multiple receptors were also reported. 14 Additionally, considering the molecular GEA subgroup, FGFR2 amplifications were more frequently shown in MSS/TP53+ or MSS/EMT subtypes per ACRG classification 8 as well as in the CIN and GS subtypes per TCGA classification.7,15
Several anti-FGFR agents have already been tested in GEA. 9 However, only recently, FGFR has been spotlighted as one of the most promising targets in GEA, thanks to positive results in the first-line setting (see below). Dovitinib, anlotinib, AZD4547, bemarituzumab and pemigatinib are the most important anti-FGFR agents, which have been evaluated in GEA in phase II and/or III trials (Figure 2). However, even though the multikinase inhibitors dovitinib and anlotinib were the first investigated in GEA, no data are available in the field.

FGFR inhibitors mechanism of action.
AZD4547 is an FGFR1-2-3 inhibitor; it was evaluated in GC in the phase II SHINE trial. 16 The trial randomized 71 metastatic GC patients with FGFR polysomy or amplification to receive paclitaxel (80 mg/m2 on days 1, 8 and 15 q28 days) or AZD4547 (80 mg bis in die (bid) for 14 days q21 days) as second-line treatment. It showed no benefit for the experimental arm in terms of progression-free survival in the entire study population (PFS: 1.8 versus 3.5 months in the AZD4547 and control arm, respectively) and the polysomy or amplified groups [hazard ratio (HR): 1.87 and 1.3, respectively]. Likewise, the benefit in OS was not statistically significant in the entire population (5.5 versus 6.6 months for AZD4547 and paclitaxel arms, respectively) and the amplified and polysomy cohorts (HR: 1.26 and 1.36, respectively). These results could be related to the high intra-tumour heterogeneity in FGFR expression. 17
Bemarituzumab is a humanized IgG1 antibody that inhibits the FGFR pathway by targeting the FGFR2b ligand-binding domain; this blocks the activation of the FGFR pathway on tumour cells and leads to cell death via antibody-dependent cytotoxicity (ADCC) by interacting with immune cells, such as natural killers. It was the first-in-class antibody against FGFR2 to be tested in GC, which showed positive results in this field. The activity of bemarituzumab was assessed in the phase II, multicentric, randomized, double-blind FIGHT trial. 18 The trial prescreened 910 patients with a new diagnosis of metastatic HER2-negative GEA for FGFR2b; of them, 275 (30.2%) had FGFR2b overexpression or FGFR2 gene amplification and were included in the analysis. One hundred and fifty-five patients were 1:1 randomized to receive chemotherapy using the FOLFOX6 schedule ± bemarituzumab (15 mg/kg q14 days and 7.5 mg/kg on day 8) as first-line treatment. FGFR positivity was reported based on immunohistochemistry (IHC) in 149/155 patients and by circulating tumour DNA (ctDNA) in 26/155. The trial demonstrated results as follows: PFS (primary endpoint): 9.5 versus 7.4 months in the experimental and control arm, respectively; HR: 0.68, p = 0.07; OS: not reached (NR) versus 12.9 months in the FOLFOX6 arm, HR: 0.58, p = 0.03; overall response rate (ORR): 53% versus 40%; the duration of response was also improved (12.2 versus 7.1 months). Those results were seen in all the subgroups; however, the best outcomes were shown in patients with higher FGFR2b expression (mOS: NR in patients with FGFR2b > 10% of tumour cells versus 11.1 months in the others, HR: 0.41; median PFS: 14.1 versus 7.3 months, HR: 0.44). The treatment was well tolerated, and the most relevant class-related toxicities mainly reported in the experimental arm were stomatitis (any grade: 31.6% versus 13%) and ocular adverse event (AE) (dry eye, any grade: 26.3% versus 6.5%). The updated results after a longer follow-up (12.5 months) confirmed those results, demonstrating a 6 months improvement in OS (19.2 versus 13.5 months) in patients in the bemarituzumab and placebo arm, respectively (HR: 0.6), with the best outcome reported in the higher FGFR2b expressers (FGFR2b > 10% of tumour cells: OS 25.4 versus 11.1 months; HR: 0.41). 19 Of note, in the trial, only 3.9% of patients had ctDNA positive and IHC negative results, suggesting that IHC (a less expensive technique than ctDNA and widely available) could be used to identify sensitive patients.
Based on these promising results, the phase III FORTITUDE-101 (NCT05052801) and FORTITUDE-102 trials (NCT05111626) are ongoing. FORTITUDE 101 (NCT05052801) assesses the efficacy of adding bemarituzumab to chemotherapy (FOLFOX6) in previously untreated advanced GEA with FGFR2b overexpression. However, given the recently published results of the Checkmate 649 trial, 20 the standard of care in the first-line treatment for HER2-negative metastatic GC is changing to include anti-programmed death-1 (PD-1) therapy. Therefore, the FORTITUDE-102 trial (NCT05111626) was designed to assess the efficacy of bemarituzumab in combination with the new standard of care (FOLFOX6 plus nivolumab) in previously untreated advanced GEA with FGFR2b overexpression. The results of those trials are awaited.
Lastly, the role of anti-FGFR agents is also being evaluated in HER2-positive GEA. In this regard, some evidence reported a role of members of the FGFR family, such as FGFR3, in acquired resistance to trastuzumab in HER2-positive GEA. 21 Based on this background, the phase II non-randomized FIGHTER trial is testing the safety and activity of pemigatinib in ⩾2nd-line treatment for metastatic HER2-positive GEA (EudraCT Number 2017-004522-14). 22
In conclusion, FGFR has become one of the most critical targets in GEA in the last few years. In particular, pending the results of FORTITUDE 101 and FORTITUDE 102, bemarituzumab may become the first FGFR2b target agent to be used in the first-line treatment for metastatic HER2-negative disease.
Claudin 18.2 in GEA: Role, inhibitors and CAR-T cell
Claudin 18.2 (CLDN18.2) is one of GEA’s most novel targets. It belongs to the family of Claudin proteins, which is made up of more than 27 proteins involved in the formation of tight cell junctions. The tight junctions regulate permeability, cell polarity, paracellular transportation and barrier functions, and transmembrane signalling.23,24 Among those proteins, Claudin 18 is mainly expressed in gastric mucosa, whereas others are typical of different kinds of tissues. 25 In general, in the gastric mucosa, Claudin 18 is present in two isoforms – Claudin 18.1 and 18.2; however, CLDN18.2 is predominantly expressed in gastric tissue, both in differentiated epithelial cells of the mucosa and in GC cells. 26 Nevertheless, in a pathological condition, such as carcinogenesis, there is an overexpression of CLDN18.2 if compared to the normal gastric mucosa, leading it to become a protein specific for GC.
In physiological conditions, CLDN18.2 is exclusively confined to the intracellular tight junctions. However, during the carcinogenesis process, the tight junctions are destroyed, leading to the exposure of CLDN18.2 to the cell surface. As a result of this process, CLDN18.2 is overexpressed on the surface of GC cells – from both primary tumours and metastases, thus being one of the most specific targets in this field with limited off-tumour on-target toxicity27,28 (Figure 1). CLDN18.2 is expressed in 70% of GCs belonging to the GS TCGA subgroup. 7 Additionally, we should consider that CLDN18.2 expression is heterogeneous in GC, showing variable levels in different locations (e.g. corpus, antrum, etc.). 29 In addition to CLDN18.2 overexpression, CLDN18.2 fusions might be present in GEA. 30 The fusions were reported mainly in diffuse (15.4% versus 1.2% in the intestinal Lauren’s subtype) or signet ring cell (SRC) GC, younger age at diagnosis (51.3 ± 12.4 years versus 60.7 ± 12.2 years) and in the females 18.5% versus 4.6%); they were linked with higher N and M stage and with poor response to chemotherapy. 27 However, the translational meaning of CLDN18.2 fusions in GEA needs further evaluation and has to be defined yet.
Regarding the role of CLDN18.2 expression as a prognostic biomarker, this is still under debate. Some evidence showed no association between CLDN18.2 positivity and outcome in GC patients, 31 while others have suggested that a loss of CLDN18.2 expression could be linked to a worse prognosis. 32 Recently, a meta-analysis evaluating the link between CLDN18.2 expression and characteristics, such as T, N, M stages, HER2 expression and Lauren’s type, showed no relation with these clinic-pathological features. 33 However, the results should be validated prospectively.
Claudin 18.2 inhibitors
Based on this background, anti-CLDN18.2 agents have been developed as potential targeted therapy in GEA. 34 Among them, zolbetuximab (also called IMAB362) is the first-in-class antibody and the most important in GEA.
Zolbetuximab is a monoclonal antibody that selectively binds CLDN18.2, leading to the activation of ADCC and complement-dependent cytotoxicity (CDC) and, thus, increasing the apoptosis of tumour cells. 35 Additionally, it has a synergistic effect when combined with chemotherapy by increasing tumour infiltrating lymphocytes in the TME, thus acting as an immunomodulator. 35
Following the results of the phase I trial, 36 zolbetuximab was tested in the phase II MONO 37 and FAST trials in GEA 38 (Table 1). In the phase IIa MONO trial, 54 patients with advanced GEA, who progressed after at least one line of systemic therapy for metastatic disease and with moderate-to-strong CLDN18.2 expression (⩾50% of tumour cells), were enrolled into three cohorts by using different dosages of zolbetuximab monotherapy (1300 mg/m2; 2600 mg/m2 and 3600 mg/m2 q14 days for five infusions). 37 The treatment showed to be active with ORR of 9% in the entire population and 14% in tumours with CLDN18.2 expression in ⩾70% of tumour cells. AEs were reported in 81.5% of patients, and nausea (61%), vomiting (50%) and fatigue (22%) were the most frequent.
Claudin 18.2 inhibitors for metastatic gastroesophageal adenocarcinomas: main phase II/III evidence and ongoing clinical trials.

State of the trial and NCT was shown only for the ongoing trials. **Preliminary results were reported for ongoing trials if available.
CLDN18.2, Claudin 18.2; EOX, epirubicin, oxaliplatin and capecitabine; HR, hazard ratio; IHC, immunohistochemistry; mOS, median overall survival; mPFS, median progression-free survival; NA, not applicable; NE, not evaluable; NR, not reported; ORR, overall response rate; SAE, serious adverse event.
The phase IIb FAST trial randomized patients affected by locally advanced, inoperable, recurrent, or metastatic and positive CLDN18.2 GEA to receive chemotherapy alone (arm 1 EOX: epirubicin, oxaliplatin and capecitabine q21 days for a maximum of eight cycles) alone or in combination with zolbetuximab (arm 2; loading dose 800 mg/m2 then 600 mg/m2) as first-line treatment. 38 Then, an exploratory third arm was added (zolbetuximab 1000 mg/m2 plus EOX). Zolbetuximab was continued as maintenance treatment in case of a stop of chemotherapy after eight cycles. In the trial, the CLDN18.2 positivity was defined as moderate (2+) to strong (3+) expression in ⩾40% tumour cells, assessed centrally by IHC. Among 730 patients initially screened, 686 tumour samples were evaluated; of these, 334 (49%) showed CLDN18.2 positivity, and 246 were randomized into the three arms. The trial showed that the addition of zolbetuximab improved PFS (primary endpoint: 7.5 versus 5.3 months, HR: 0.44, 95% confidence interval (CI): 0.29–0.67; p < 0.0005), OS (13 versus 8.3 months, HR: 0.55; 95% CI: 0.39–0.77; p < 0.0005) and ORR (39% versus 25%, p: 0.034), especially in patients with higher CLDN18.2 expression (⩾70% of tumour cells; HR: 0.38; 95% CI: 0.23–0.62; p < 0.0005). The addition of zolbetuximab did not increase AEs compared to chemotherapy; low-intensity nausea and emesis (mainly Grades 1 and 2) were the most common (81.8% and 67.5%, respectively). Of note, the subgroup analysis showed that the benefit of adding zolbetuximab was higher in diffuse GC if compared to intestinal one in PFS and OS. The analysis regarding the quality of life showed no difference between arms during the entire treatment, with an improvement in the scores during the maintenance period with zolbetuximab alone. 44 However, we should consider some aspects in the interpretation of those data. In particular, even if the FAST trial showed positive results, it was conducted between 2012 and 2014, using a triplet epirubicin-based chemotherapy as a control arm that is not considered the standard of care in the first-line setting anymore. Additionally, the control arm performed worse than the historical data by using the epirubicin-based triplet. Lastly, the strong CLDN18.2 expressers might drive the benefit reported in the trials.
Subsequently, the phase II non-randomized ILUSTRO trial was conducted (NCT03505320). The trial evaluates the activity and safety of zolbetuximab plus chemotherapy (FOLFOX6) as first-line treatment for 21 GEA patients with HER2-negative and high CLDN18.2 expression (⩾75% of tumour cells). The preliminary results showed a median PFS of 13.7 months (95% CI: 7.4–not estimable), ORR 63.2% (95% CI: 38.4–83.7); the most frequent grade 3/4 AEs were decreased neutrophil count and neutropaenia (33.3% and 28.6%, respectively). 39 However, the trial is still ongoing, and definitive results are awaited.
Additionally, the phase III trials SPOTLIGHT (FOLFOX6 ± zolbetuximab as first-line treatment for metastatic HER2-negative GEA; NCT03504397) 40 and GLOW (Xelox ± zolbetuximab in the same setting; NCT03653507) are currently running 42 as well as studies exploring the combination of anti-CLDN18.2 agents and immunotherapy (NCT03505320). 43 The preliminary results of the SPOTLIGHT trial were recently presented. 41 The trial enrolled 566 patients with CLDN18.2-positive (CLDN18.2 expression in ⩾75% of tumour cells), HER2-negative, locally advanced unresectable or metastatic GEA. It met the primary endpoint, showing improvement in both PFS (primary endpoint) and OS for the experimental arm (median PFS: 10.61 versus 8.67 months in the experimental and control arm, respectively, HR: 0.75, p = 0.006; median OS: 18.23 versus 15.54 months, HR: 0.75, p = 0.005). However, the addition of zolbetuximab to chemotherapy did not improve ORR (60.7% versus 62.1%). Additionally, although the trial reports significant results, the clinical relevance is modest if we look at HRs and absolute improvements. There was a higher rate of nausea, emesis and decreased appetite in the experimental arm (respectively, 81.0% versus 60.8%; 64.5% versus 34.5%; 47% versus 33.5%); however, the rate of serious events was similar (44.8% versus 43.5% in the FOLFOX6+ zolbetuximab and FOLFOX6 arm, respectively).
Even if the description of the role and use of immune checkpoint inhibitors in GEA was not in the aim of our narrative review, based on those results, there possibly are two treatment strategies in first-line for metastatic programmed death ligand 1 (PD-L1) according to combined positive score (CPS) ⩾ 5 GC: chemotherapy in combination with nivolumab or with zolbetuximab. However, at the time of writing no direct comparison exist and the choice between those two options is strictly related to PD-L1 and CLDN18.2 expression and to local approval.
Lastly, CLDN 18.2 antibody drug conjugate, such as CMG901 and CPO102, are currently in early-phase development for solid tumours, including GEA (NCT04805307 for CMG901 and NCT05043987 for CPO102), representing the future perspectives in this field.
Claudin 18.2 CAR-T cells
Inspired by the outstanding results of chimeric antigen receptor (CAR)-T cell-based therapy in haematological malignancies, 45 research is now focusing on its use also in solid tumours, 46 including GC. In brief, CAR-T cells are derived from T cells isolated from patients’ blood; those cells are modified by a viral vector introducing the CAR, which can recognize a tumour-associated antigen (TAA). In the end, the modified T cells are infused into the patient with the new ability to recognize TAA on the surface of cancer cells. This binding leads to cytotoxicity by T cell immune-mediated mechanisms (Figure 3(a)). Additionally, it was shown that CAR-T cell therapy could be considered a treatment with long-term effects, mainly due to the persistence of CAR-T cells, especially in the TME. 47 Considering the high specificity of CLDN18.2 in GC cells and its exposure to the cell surface, CLDN18.2 was recognized as one of the best targets for CAR-T cells. However, several other targets are also under evaluation for developing CAR-T cell treatments in GC, including HER2 and Mucin 1 (MUC1), for example. 48 Nevertheless, considering the reported AE, such as cardiotoxicity, improvements in the safety profile of anti-HER2 CAR-T cells are needed, and trials are ongoing.

Cellular therapies in GEA: CAR-T cell (a) and SPEAR-T cell (b) processes.
For CLDN18.2 in GEA, no phase II trial about CLDN18.2-targeted CAR-T cell exist at the time of writing. Thus, even if we assumed to include only phase II/III trials in this review, we decided to report here the early-phase results, which is the highest level evidence in this field. In this regard, the recently published interim analysis results of the ongoing, open-label, single-arm, phase I trial using a CLDN18.2-targeted CAR-T cell (CT041) are the most important. 49 The trial included 59 patients with advanced gastrointestinal tumours, who progressed after at least one line of standard systemic treatment, with CLDN18.2 expression by IHC (⩾2 and positive tumour cell rate ⩾40%); of them, 49 patients were treated, and 37 patients were included in the interim analysis (28 patients with HER2-negative GEA). In brief, the patients received apheresis followed by bridging chemotherapy and preconditioning therapy with a low dose of nab-paclitaxel plus fludarabine/cyclophosphamide during the manufacturing of CT041. Following this, CT041 was infused back into the patients. The treatment was well tolerated, and no dose-limiting toxicities were reported, whereas all patients had haematological AEs due to bridging chemotherapy and the preconditioning phase. In the GEA cohort, the treatment showed to reach an ORR of 57.1% and disease control rate (DCR) of 75%, with the best results in patients with Lauren intestinal tumours (70% ORR) or high CLDN18.2 expression, defined as 2+ and 3+ at IHC in ⩾70% of tumour cells. Also, outcomes were similar for patients previously treated with and without taxanes or anti-programmed PD-1 and PD-L1 agents. The median PFS in the entire GC cohort was 4.2 months and the OS rate at 6 months was 81.2%. Nevertheless, the PFS is shorter than that in the haematological malignancies, perhaps related to the shorter persistence of CT041 in the blood (28 days). However, few patients with undetectable CT041 had durable responses, and the evaluation of the persistence of CT041 in solid tumour tissues is hard to perform, so future investigations are needed in this regard. Of note, in the case of patients with peritoneal disease, the concentration of CT041 was higher in the ascites than in the blood, suggesting a possible role of CT041 in the treatment of peritoneal carcinosis. Additionally, the CLDN18.2 expression did not change due to CT041 treatment, suggesting that there will be mechanisms of acquired resistance other than antigen escape in this case and representing a first few steps in the research about resistance mechanisms in this field.
However, even if CAR-T cell therapy could be considered intriguing and a sort of ‘dream’ from an oncologist’s point of view (highly specific and targeted therapy), its use has some limitations. First, developing personalized CAR-T cells is a long and expensive process. Additionally, the heterogeneity of tumour cells, the persistence of CAR-T cells in tumour tissue and the complex interactions between tumour cells and TME could lead to the appearance of drug resistance due to the lack of specificity of CAR-T and/or to lack of function due to the immunosuppressive environment. 50 Therefore, despite promising results, further trials are needed to confirm CAR-T cell-based therapies’ role and introduce them in the clinical practice for GEA.
Then, BiTE® (bispecific T cell engager) antibody which specifically links two molecular targets at the same time, such as CLDN18.2 and CD3, for example, are also in development in the GC field (NCT04260191). 51
Briefly, BiTE® enhance the link between endogenous T cells to tumour-expressed antigens without genetic manipulation of T cell or ex vivo expansion by recognizing a tumour-specific target, such as CLDN18.2, and a T cell-specific target, such as CD3. This bispecific link leads to T cell activation and immune response against tumour cells (cytotoxic effect). Interestingly, based on this mechanism of action, any T cell can be involved without co-stimulators. 52
Another cellular therapy using the TCR receptor on T cell (so-called SPART cell) is developing in GC. In particular, SPEAR-T cells are engineered T cells derived from patients and able to identify and bind a specific human leukocyte antigen (HLA). The binding leads to recognizing specific cancer cell antigens to destroy cancer cells and decrease systemic toxicity (Figure 3(b)). In this context, the phase II SURPASS-2 trial assesses the safety and efficacy of SPEAR-T cell targeting MAGE-A4 antigen in HLA-A*02 positive patients with advanced GEA after the failure of one line of systemic treatment at least (NCT04752359). 53
Antiangiogenetic agents
In general, angiogenesis plays a crucial role in different processes involved in cancer development, such as drug diffusion, anaerobic metabolism, immune dysfunction and spread of metastases, and so on and in several kinds of solid tumours, including GEA. Thus, the inhibition of neo-angiogenesis has been recognized as a mainstream antitumoral effect, and several pharmaceutical compounds have been developed over the last decades to target that. However, most antiangiogenetic agents, such as bevacizumab, failed to significantly improve outcomes in GEA patients. 54
However, almost 10 years ago, the phase III trials REGARD 55 and RAINBOW 56 showed positive results by using ramucirumab in GEA; following those results, ramucirumab is the only antibody targeting VEGFR-2 approved in clinical practice as the second-line treatment for metastatic disease. 5 Those results were also confirmed in some real-life experiences, including not selected patients57,58 and patients with HER2-positive metastatic GEA after the failure of first-line treatment. 59
However, after that ‘golden era’ for antiangiogenesis in GEA, the subsequent phase III RAINFALL trial did not show improvement in the outcomes in first-line treatment for HER2-negative metastatic GEA by using ramucirumab in combination with cisplatin-5fluorouracil, perhaps also due to the choice of this chemotherapy backbone. 60 Recently, the phase II RAMIRIS trial results were published. 61 The trial randomized 110 patients with metastatic GEA candidate to a second-line treatment to receive ramucirumab in combination with paclitaxel (standard of care) or FOLFIRI (experimental arm). The trial did not meet the primary endpoint (6-month OS rate ⩾65%), showing a 6-month OS rate of 54% in the FOLFIRI plus ramucirumab arm (95% CI: 44−67%), no differences in OS (HR: 0.97, 95% CI: 0.62–1.52); the safety profile was also comparable between the two arms. However, PFS and ORR improved in the experimental arm (PFS: HR: 0.73; ORR: 22% versus 11%), which may be related to the fact that FOLFIRI plus ramucirumab performed better in the subgroup of patients who already have received docetaxel as previous treatment (HR: 0.49; ORR: 25% versus 8%). Thus, even if the trial was formally negative, these results could pave the way for further evaluations in this subgroup of patients.
Nowadays, there are two main innovative areas of research in the anti-angiogenetic field: one exploring the role of predictive biomarkers for response to treatment and the other finding antiangiogenetic agents beyond ramucirumab to improve the prognosis of GEA patients, as a single agent or in combination with immunotherapy. Regarding the first point, although antiangiogenics – specifically ramucirumab – are targeted treatments, up to date, there are no biomarkers available, and, thus, the landmark trials did not include selected patients. A recently published prospective study evaluated serum biomarkers in 35 patients receiving paclitaxel and ramucirumab as second-line treatment. 62 The analysis showed that patients who reported a response to treatment had a lower level of vascular endothelial growth factor C (VEGFC) and angiopoietin-2; otherwise, those two markers were higher in case of progression of the disease, suggesting a possible role of VEGFC and angiopoietin-2 as predictive biomarkers of response to antiangiogenic in GEA.
Regarding the second point, the extensive discussion about early-phase combinations between target agents and immunotherapy is beyond the scope of this review. However, in this context, regorafenib and lenvatinib are the most important multikinase inhibitors with antiangiogenetic activity explored in GEA (Table 2).
Novel antiangiogenic for metastatic gastroesophageal adenocarcinomas: main phase II/III evidences and relevant ongoing clinical trials.

State of the trial and NCT was shown only for the ongoing trials. **Preliminary results were reported for ongoing trials if available.
mOS, median overall survival; mPFS, median progression-free survival; ORR, overall response rate; NA, not applicable; NR, not reached; HR, hazard ratio.
Regorafenib targets VEGFR1-2, PDGFRβ, RAF, RET and KIT; its role in GEA was explored in the open-label, phase II INTEGRATE trial. 63 The study randomized 2:1 a total of 152 pretreated patients with metastatic GEA (⩾2nd line) to receive regorafenib 160 mg daily (days 1–21 q28) or best supportive care. Regorafenib was associated with an improvement in median PFS in all the subgroups (2.6 months (95% CI: 1.8–3.1 months) versus 0.9 months (95% CI: 0.9–0.9); HR: 0.40 (95% CI: 0.28–0.59, p: 0.001); a favourable trend in OS for the experimental arm (5.8 versus 4.5 months; HR: 0.74; p = 0.147) was also reported. However, a crossover was allowed, affecting the results. Based on these data, the phase III INTEGRATE-II trial was conducted and the results were recently presented. 64 The trial randomized 251 metastatic and heavily pretreated GEA patients (⩾2 prior lines of therapy with a platinum + fluoropyrimidine) to receive regorafenib or placebo (2:1 randomization). The trial showed a median OS of 4.5 versus 4 months in the regorafenib and placebo arm, respectively (HR: 0.70; p = 0.011), and median PFS of 1.8 versus 1.6 months (HR: 0.52; p ⩽ 0.0001). The safety profile of regorafenib was reported to be similar to previous reports, but not shown in the abstract. Nevertheless, we should take into account that, although the trial reports significant results, the clinical relevance is modest if we look at HRs and absolute improvements.
Additionally, the role of regorafenib is being explored as combined therapy with other agents. In this context, the early Ib phase REGONIVO trial showed promising results in GEA by combining regorafenib with immunotherapy, exploiting the capacity of regorafenib to modulate TME and tumour-associated macrophages.71,72 Then, the recently published phase Ib/II REPEAT trial assessed the tolerability and activity of regorafenib and paclitaxel as second-line treatment for metastatic GEA patients. 65
The trial showed promising results, with median OS and PFS of 7.8 and 4.2 months, respectively, however without significant improvements if compared to paclitaxel plus ramucirumab in a propensity score matched cohort (OS: p = 0.08 and PFS: p = 0.81). The exploratory analysis regarding biomarkers showed a link between increased circulating levels of galectin-1 and chromosome 19q13.12-q13.2 amplification and poor prognosis.
Lastly, lenvatinib, which targets VEGFR1-3, FGFR1-4, PDGFRα and RET, is the most important multikinase inhibitor beyond regorafenib to be tested in GEA, mainly in combination with immunotherapy. The phase II EPOC1706 evaluated the activity of lenvatinib plus pembrolizumab in 29 metastatic GEA patients in Japan (first- and second-lines). 66 The combination showed promising activity with 69% ORR and a good safety profile.
The phase II LEAP-005 trial (NCT03797326) is a non-randomized, open-label study to explore the efficacy and safety of lenvatinib plus pembrolizumab as ⩾third-line treatment in patients with solid tumours, including 31 GC patients. 67 The preliminary results reported ORR: 10%, DCR: 48%, median PFS: 2.5 months, median OS: 5.9 months; 90% and 42% were the all-grades AE rates and grade 3–5 rates, respectively (one patient died due to haemorrhage). The trial is currently ongoing after the expansion of the GC cohort, and definitive results have to come yet. Then, several trials testing lenvatinib in GEA are ongoing, including the phase III LEAP-015 trial (pembrolizumab, lenvatinib, chemotherapy in first-line for advanced GEA, NCT04662710).68,69 The results of the safety run-in analysis for LEAP-015 were recently presented: 15 patients received at least one dose of experimental combination; 93% of them had AEs, and 53% were grade 3/4. No grade 5 and no grade ⩾3 immune-mediated AEs or infusion reactions occurred. 70
In conclusion, angiogenesis is crucial in cancer development and progression. Ramucirumab is the principal agent with antiangiogenetic activity currently used in GEA; however, several trials with other antiangiogenic are ongoing in this field, especially in combination with immune-oncological agents.
New anti-HER2 agents
HER2 has been extensively studied in GEA, representing the first receptor targeted successfully in those tumours. 73 In particular, according to international guidelines and based on the landmark ToGA trial, 74 trastuzumab has been the unique target agent approved as the standard of care in the first-line treatment for a long time for HER2-positive GEA. 5 Nevertheless, subsequent trials with other anti-HER2 drugs failed to improve the outcomes for GEA patients. 54 For a detailed historical description of anti-HER2 agents and the results of trials investigating the role of the combination of immunotherapy and anti-HER2 inhibitors, we suggest referring to the existing literature.6,54,75
Antibiotic-drug conjugates and monoclonal and bispecific antibodies are the newest class of anti-HER2 target drugs investigated in GEA. T-Dxd is the most important in the first category. Briefly, T-Dxd is made up of an anti-HER monoclonal and humanized antibody bound to a cytotoxic payload (topoisomerase I inhibitor). A key characteristic of T-Dxd is the capacity to diffuse through the cell membrane of targeted cells, leading to cytotoxicity in the surrounding area of non-HER2-positive cells. This process is known as the bystander effect. 76 This feature is important in GEA, where HER2 expression is heterogeneous and dynamic.77–79 Following the promising phase I trial results, 80 the phase II DESTINY-Gastric01 trial was conducted in Asian patients. 81 The trial randomized 187 heavily pretreated patients with metastatic HER2 expressed GEA to receive 2:1 either T-Dxd (6.4 mg/kg q21 days) or chemotherapy according to physician’s choice (irinotecan or paclitaxel monotherapy) as ⩾third-line treatment. All patients have already received a previous line of treatment including trastuzumab. The trial showed that T-Dxd improved the outcomes with ORR of 51% versus 14% in the control arm (complete responses 9% versus 0%) (p < 0.001); median OS: 12.5 versus 8.4 months (HR: 0.59; p: 0.01), median PFS: 5.6 versus 3.5 months (HR: 0.47). Most responses were recorded according to HER2 expression (58% in HER2 3+ versus 29% in the HER2 2+/FISH+ subgroup). Haematological grade 3–4 AEs were the most frequent (decreased neutrophil count: 51% versus 24%; anaemia: 38% versus 23%, decreased white-cell count: 21% and 11%). Additionally, interstitial lung disease or pneumonitis was reported as T-Dxd specifically drug-related AE (10% versus 0%, 9/12 patients had grade 1–2 AE). Of note, the trial enrolled patients with HER2 expression, which means HER2 high (also known as HER2-positive) and HER2 low (1+ at IHC or 2+ IHC/FISH negative); however, the first analysis only included HER2-positive patients.
Subsequently, the HER2-low cohort results were published. 82 The trial included 21 patients with HER2 2+/FISH negative tumours (cohort 1) and 24 patients with HER2 1+ tumours (cohort 2); those patients were naive to anti-HER2 treatment as per international guidelines 5 and received T-Dxd as ⩾third-line treatment. There was an ORR of 26.3% and 9.5% for the experimental arm in cohorts 1 and 2, respectively; also, in this case, major responses were recorded (partial responses in 5 and 2 patients, respectively). T-Dxd also improved survival outcomes: median OS: 7.8 and 8.5 months in cohorts 1 and 2, respectively; median PFS: 4.4 and 2.8 months. The safety analysis confirmed the AEs already reported in the HER2-positive cohort. Based on these results, T-Dxd showed to be active also in patients with heavily pretreated HER2 low GEA, which is an entirely new concept in the field of GEA. Although the trial involved only a small number of patients and was entirely conducted in Asia, these results pave the way for further phase III investigations, following the example of breast cancer. 83
In Western patients, the phase II non-randomized DESTINY-Gastric02 trial was conducted (NCT04014075). The trial included 79 patients with HER2-positive metastatic GEA who progressed to trastuzumab; they received T-Dxd as ⩾second-line treatment. The recently presented updated results recently demonstrate a centrally confirmed ORR of 41.8%, median OS: 12.1 months, median PFS: 5.6 months, AEs: 100% (grade ⩾3: 55.7%; nausea, vomiting and fatigue were the most frequent (67.1, 44.3 and 57%, respectively); interstitial lung disease: 10.1%). 84 Lastly, combinations with T-Dxd and chemotherapy in GEA are also being tested in ongoing trials, such as DESTINY-Gastric 03 (NCT04379596). 85 Taking all these results together, recently, the European Medication Agency recommended an extension of therapeutic indications for T-Dxd, adding ‘monotherapy for the treatment of adult patients with advanced HER2-positive GEA who have received a prior trastuzumab-based regimen’. 86
Regarding other monoclonal antibodies, margetuximab has been explored in HER2-positive GEA, mainly in combination with immunotherapy. Margetuximab is an FC-engineered antibody against HER2 with a high affinity for CD16A, which is expressed in many kinds of immune cells (natural killer, T cells, dendritic cells, macrophages and monocytes). This could lead to a modulation of both innate and adaptive immunity. 87 The phase II/III MAHOGANY trial evaluated the safety and efficacy of the chemotherapy-free regimen of margetuximab and retifanlimab (anti-PD-1) as first-line treatment for HER2-positive GEA; the trial was designed to start with a cohort A (patients with HER2 IHC3+, MSS and PD-L1 CPS ⩾ 1% by IHC) with safety and ORR as primary objectives. 88 It included 43 patients in the part 1 of that cohort; it showed 53% ORR, 73% DCR and a good safety profile (grade 3 AEs: 18.6%). However, following the recent advances in therapeutics in this field, showing positive results by using chemo-immunotherapy in the first-line setting for HER2-positive GEA, the sponsor decided not to continue with the development of the MAHOGANY combination (chemo-free) and the trial did not move to the next phase.
Zanidatamab (also known as ZW25) is the most important bispecific IgG1 antibody studied in GEA; it binds both the extracellular (ECD4) and the dimerization domain (ECD2) of HER2, leading to the downregulation of HER2 as well as to ADCC, CDC and phagocytosis. Zanidatamab was evaluated in an open-label, single-arm, phase II trial as a combined first-line treatment (with Xelox or platinum and 5fluorouracil). The study included 28 patients with advanced/metastatic HER2-positive GEA and showed promising activity for the combination as preliminary results (confirmed ORR: 75%, median PFS: 12 months). 89 Other trials in this field, including phase III HERIZON-GEA-01, 90 are ongoing (NCT04276493, NCT05270889, NCT03929666).
Lastly, even if we decided to focus on novel anti-HER2 agents in this review, the Keynote-811 trial is worth a brief mention due to its promising interim results. 91 The double-blind, global, phase III trial randomized 434 patients affected by metastatic HER2-positive GC to receive chemotherapy plus trastuzumab ± pembrolizumab 200 mg flat dose every 3 weeks as first-line treatment. Of note, in this trial, chemotherapy was according to ToGA schedule (cisplatin plus fluorouracil) or capecitabine and oxaliplatin; this was the first time a chemotherapy backbone was different from the ToGA one in this setting. The interim analysis demonstrated 22.7% improvement in ORR (74.4% versus 51.9%, respectively), 6.9% in DCR (96.2% versus 89.3% and 8.2% in complete responses in the experimental arm (11.3% versus 3.1%). Additionally, the responders had a long-term response. The addition of pembrolizumab to chemotherapy was safe, and the toxicities were comparable in both arms (grade ⩾ 3 AE: 57.1% versus 57.4% in the experimental and control arm, respectively; serious AE: 31.3% versus 38.4%; immune-related AE: 33.6% and 20.8%). Of note, the trial did not select patients according to PD-L1; however, stratification according to PD-L1 CPS was done, and 84% of patients had a PD-L1 CPS ⩾1. These results suggest a possible role for the combination of pembrolizumab and chemotherapy plus target therapy in HER2-positive tumours in the future; however, final results about efficacy are awaited before changing the standard of care in this field.
In conclusion, even after several disappointing results following the ‘first-in GEA’ ToGA trial, HER2 remains an actual and appealing target in GEA.
Future perspectives and conclusion
GEA is a complex disease with a poor prognosis in most cases. However, a scenario with new molecular targets in GEA is evolving, and the molecular mechanisms driving tumour development and progression still need to be understood entirely. Our lack of understanding led to the failure of several clinical trials in the field over the last decades. 54 If this is not enough, we should consider the huge heterogeneity of GEA, which can complicate the picture. In particular, it can be distinguished as spatial and temporal heterogeneity; then, among the first type, we should consider the heterogeneity within a patient (intra- and inter-tumour heterogeneity) or between patients.92,93 Additionally, we should consider that tumours are dynamic, showing different molecular profiles over time and under the selective pressure of treatments. So, in attempting to develop and choose a more and more personalized treatment for each patient – so-called precision medicine – the genomic heterogeneity can be considered a chance.
However, a dynamic tumour genomic profiling could guide to tailor the treatment in GEA by understanding the molecular peculiarities driving cancer ‘here and now’. Then, performing a new tumour biopsy at the time of progression became extremely important. In this regard, the PANGEA, 94 the VIKTORY Umbrella 95 and K-Umbrella GC (NCT02951091) 96 trials paved the way for future horizons in GEA. In particular, the phase II PANGEA trial stratified and treated metastatic GEA patients according to their own tumour’s molecular characteristics: MSI-high, PD-L1 CPS >10, tumour mutational burden count, EBV positive, HER2-positive, epithelial growth factor receptor (EGFR) amplified, FGFR2 amplified, MET amplified, RAS-like, EGFR expressing, all-negative. The trial showed promising results as follows: 1-year OS rate of 69.4% (p < 0.001), median OS: 16.4 months. 94 Of note, the study underlined the impact of spatial (primary site versus metastasis – synchronous or metachronous) and temporal (over time) heterogeneity. Likewise, the VIKTORY Umbrella trial treated 715 metastatic GEA patients in second-line treatment according to one of the following eight categories: RAS aberration, TP53 mutation, PIK3CA mutation/amplification, MET amplification, MET overexpression, all-negative, TSC2 deficient, or RICTOR amplification. 95 The trial showed a benefit in the case of personalized treatment if compared to the standard of care regardless of molecular profiling (median OS: 9.8 versus 6.9 months, p < 0.0001; median PFS: 5.7 versus 3.8 months, p < 0.0001). Recently, the first reports from the ongoing K-Umbrella GC study were shown. 96 The trial randomized 329 HER2-negative metastatic GC patients candidate for second-line treatment into four groups: EGFR 2+/3+, PTEN loss/null, PD-L1+, deficient mismatch repair proteins (dMMR)/MSI-high, or EBV-related cases; all-negative. It showed exciting results (median PFS and OS were 3.8 and 8.9 months in the experimental-biomarker-guided arm, respectively, versus 4.1 and 8.7 months in the control arm) with a worse prognosis in the case of PTEN loss/null presence.
Then, in this review we focused on the most important and advanced findings in terms of novel target agents for GEA. However, other potential molecular targets, such as tumour growth factor beta, MET and the compounds of TME are worthy of further exploration in this field.
Lastly, the methodology used to evaluate GEA molecular landscape and tailor the treatment is evolving. In particular, IHC can be routinely used to assess the FGFR2, CLDN18.2, HER2, MMR and PD-L1, whereas the NGS is helpful as a complementary method and in case of co-expression of mutations/amplification. 97
In conclusion, even if GEA is a challenge for each oncologist, the research and knowledge regarding the molecular profile and novel targets are improving to overcome the barriers to developing personalized approaches.