
Abstract
Gastric cancer (GC) is one of the most common malignancies worldwide. The histology- and morphology-based Lauren classification of GC has been widely used for over 50 years in clinical practice. The Lauren classification divides GC into intestinal and diffuse types, which have distinct etiology, molecular profiles, and clinicopathological features. Diffuse-type GC (DGC) accounts for approximately 30% of GCs. Tumor cells lack adhesion and infiltrate the stroma as single cells or small subgroups, leading to easy dissemination in the abdominal cavity. Clinically, DGC has aggressive traits with a high risk of recurrence and metastasis, which results in unfavorable prognosis. Although systemic chemotherapy is the main therapeutic approach for recurrent or metastatic GC patients, clinical benefits are limited for patients with DGC. Therefore, it is urgent to develop effective therapeutic strategies for DGC patients. Considerable research studies have characterized the molecular and genomic landscape of DGC, of which tight junction protein claudin-18 isoform 2 (CLDN18.2) and fibroblast growing factors receptor-2 isoform IIIb (FGFR2-IIIb) are the most attractive targets because of their close association with DGC. Recently, the impressive results of two phase II FAST and FIGHT trials demonstrate proof-of-concept, suggesting that anti-CLDN18.2 antibody (zolbetuximab) and FGFR2-IIIb antibody (bemarituzumab) are promising approaches for patients with CLDN18.2-positive and FGFR2-IIIb-positive GC, respectively. In this review, we summarize the clinicopathological features and molecular profiles of DGC and highlight a potential therapeutic target based on the findings of pivotal clinical trials.
Introduction
Gastric cancer (GC) remains a major clinical challenge. It is the fifth most common cancer and the third leading cause of cancer-related deaths worldwide. 1 The incidence of GC is high in East Asia, Eastern Europe, and South America. Helicobacter pylori infection is closely associated with the development of GC, and its eradication is effective in reducing GC incidence. 2 However, because of the lack of early clinical signs, GC is still frequently diagnosed at advanced stages that are not amenable to curative resection. For such patients, systemic chemotherapy is the main therapeutic option for prolonging survival and improving symptoms and quality of life.3,4
The Lauren classification divides GC into intestinal and diffuse types based on cell histology and morphology and is clinically well used because of different phenotypes, responses to treatment, and prognoses. 5 Diffuse-type GC (DGC) cells tend to scatter noncohesively into the stoma of the stomach and disseminate easily in the abdominal cavity. 5 In addition, DGC cells have enhanced invasive abilities in the stomach wall and lymphatic vessel compared with intestinal-type GC (IGC) cells. 6 Consequently, aggressive phenotypes of DGC result in poor survival outcomes via peritoneal dissemination or lymph node metastasis,6–8 and high recurrence frequency after curative surgery. 9 DGC accounts for approximately 30% of GCs and is trending toward increasing prevalence.5,7 Eradication of H. pylori may induce an increased risk of developing DGC, in contrast to IGC. 10 There is an urgent need to develop effective therapeutic strategies to overcome poor tumor cellularity in DGC.
Many molecular-targeted agents have failed to demonstrate significantly improved overall survival (OS) in clinical trials for patients with recurrent or metastatic GC, partially due to a lack of selective biomarkers and/or intratumoral heterogeneity. Currently, human epidermal growth factor receptor 2 (HER2),11,12 vascular endothelial growth factor receptor 2 (VEGFR2),13,14 and programmed death-1 (PD-1)15,16 are clinically validated targeted molecules in GC. However, these molecular-targeted agents may have limited clinical utility for patients with DGC because of the rare frequency of targeted molecule aberrations and weak efficacy. There is also less benefit from chemotherapy in DGC. 17 An in-depth understanding of the complexity and diversity of molecular profiles will pave the way for establishing personalized molecular-targeted medicine for DGC patients.
Based on The Cancer Genome Atlas (TCGA) molecular classification, GC can be categorized into four subtypes: microsatellite instability (MSI), Epstein–Barr virus (EBV)-positive, chromosomal instability (CIN), and genomically stable (GS) tumors. 18 GS tumors have frequent fusions of tight junction protein claudin-18 (CLDN18), and mutations of cadherin 1 (CDH1) or ras homolog family member A (RHOA), which mediates epithelial disintegration and diffuse-type phenotype.18–21 In addition, comprehensive molecular analyses demonstrate the aberration of fibroblast growing factor receptor-2 (FGFR2) as a critical molecule in DGC. 18 Recently, promising results of anti-CLDN18 isoform 2 (CLND18.2) antibody, zolbetuximab, and anti-FGFR2 isoform IIIb (FGFR2-IIIb) antibody, bemarituzumab, were shown in phase II FAST 22 and FIGHT 23 trials, respectively. Thus, CLDN18.2 and FGFR2-IIIb are relevant therapeutic targets and have attracted considerable attention as new hope for DGC patients.
In this review, we summarize the biology, molecular, and genetic landscape and clinical features of DGC, and highlight a potential therapeutic target based on the findings of pivotal clinical trials.
Clinicopathological and molecular features of diffuse-type gastric cancer
The histology- and morphology-based Lauren classification of GC has been widely used for 50 years. 5 According to the Lauren classification, GC is mainly divided into two types (IGC and DGC), which have distinct etiology, clinicopathological features, and molecular profiles.
Clinicopathological features of diffuse-type gastric cancer
Clinicopathological features according to the Lauren classification are shown in Table 1. IGC is characterized by cohesive tumor cells with a glandular or intestinal structure, leading to an expanding growth pattern, while DGC is characterized by poorly cohesive tumor cells with few or no glandular structures, leading to a diffusely infiltrating growth pattern. DGC cells induce fibrosis and nestle within a rich fibrous stroma, which easily spreads not only along the stomach wall, but also in the upper layers of the stomach wall.24,25 In a meta-analysis of GC transcriptome data integrating 940 gastric transcriptomes, DGC showed higher stromal gene expression profiles associated with extracellular matrix biology and stromal cells than IGC, and DGC patients with high stromal profiles had more aggressive tumor biology and poor prognostic outcomes compared with those with low profiles. 26
Clinicopathological features according to the Lauren classification.
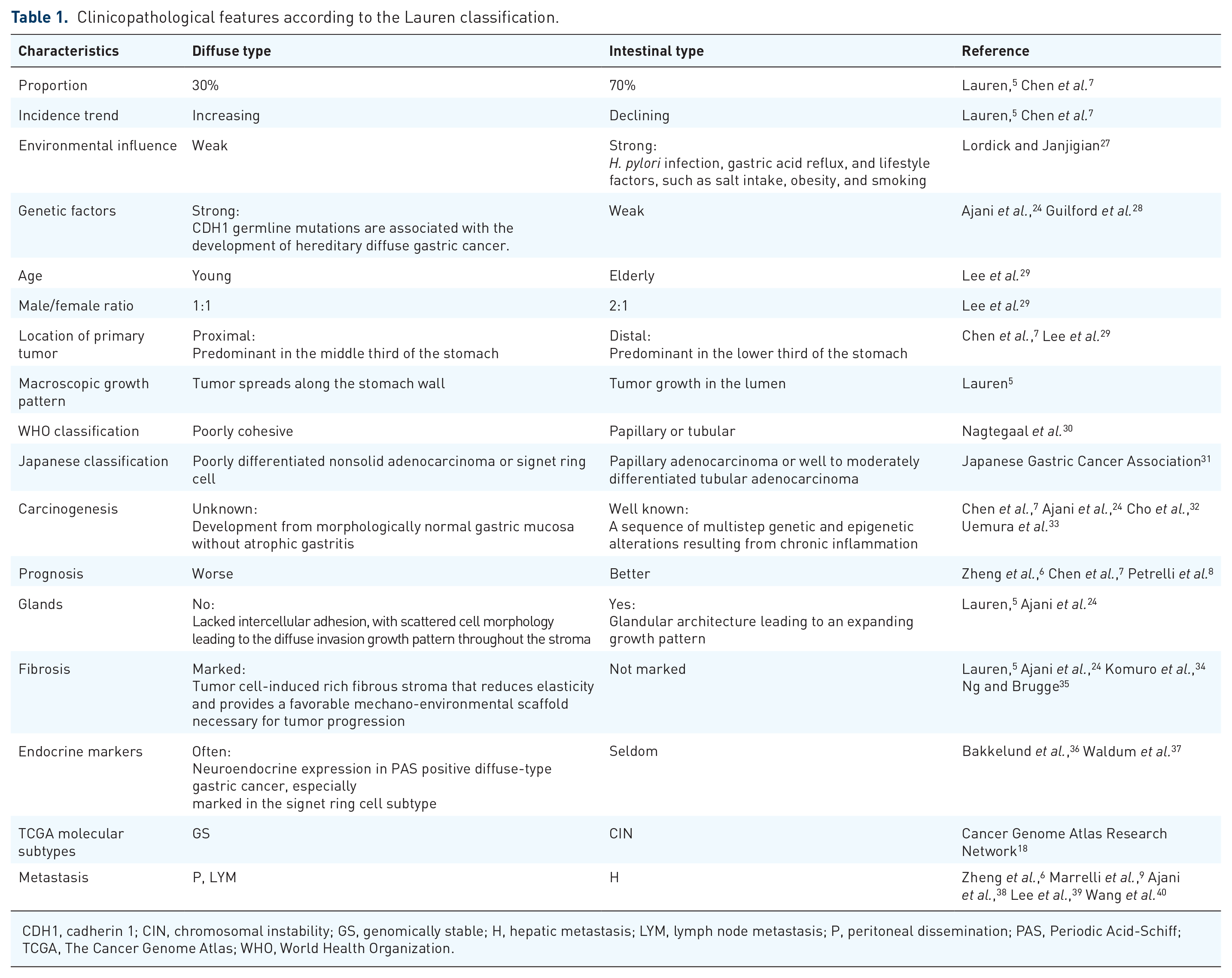
CDH1, cadherin 1; CIN, chromosomal instability; GS, genomically stable; H, hepatic metastasis; LYM, lymph node metastasis; P, peritoneal dissemination; PAS, Periodic Acid-Schiff; TCGA, The Cancer Genome Atlas; WHO, World Health Organization.
DGC is more prevalent in younger, larger tumors that are infiltrating, proximal stomach predominant, advanced stage, advanced in depth of invasion, and poorly differentiated, scirrhous type stromal reactions, and lymphovascular invasions.7,27,41 Clinically, the two histology types of GC have different patterns of metastatic spread and prognostic outcomes. DGC tends toward peritoneal dissemination, lymph node metastasis, and direct extension into neighboring tissues, while IGC is associated with hematogenous metastasis including liver.6,9,38–40 DGC has a high risk of peritoneal recurrence, even after curative surgery. 9 In a total of 20,218 patients from the Surveillance, Epidemiology, and End Results database, DGC patients had significantly less favorable cancer-specific survival compared with IGC patients, with a hazard ratio (HR) of 1.44 [95% confidence interval (CI), 1.38–1.50]. In a large cohort study of 3071 patients who underwent gastrectomy, DGC was an independent prognostic factor. 7 A meta-analysis including 61,468 GC patients demonstrated similar unfavorable OS of patients with DGC versus IGC in all patients (HR, 1.23; 95% CI, 1.17–1.29) and in patients treated with adjuvant therapy only (HR, 1.27; 95% CI, 1.17–1.37). 8 Collectively, DGC cells have aggressive traits and a high risk of recurrence and metastasis, which results in poor prognosis.
Molecular classification of diffuse-type gastric cancer
GC is a heterogeneous disease with diverse genetic and molecular levels, and four molecular subtypes (MSI, EVB, CIN, and GS tumors) were defined according to the results of comprehensive molecular analyses in the TCGA project.18,42 The GS subtype often shows a diffused cellular morphology due to the frequent loss of cell adhesion-related molecules via CLDN18-Rho GTPase-activating protein-6 or 26 (ARHGAP6, ARHGAP26) fusions (15% of GS subtype), mutations of CDH1 (37%), and RHOA (15%).18–21 In fact, DGC predominantly belongs to the GS subtype, and 73% of GS subtypes in the TCGA cohort were DGC. 18 These genetic alterations are mutually exclusive, suggesting a contribution to carcinogenesis depending on cell adhesion-related signaling in DGC.
The Asian Cancer Research Group (ACRG) proposed four molecular subtypes according to the distinct molecular profiles and clinical outcomes using gene expression profiling, genome-wide copy number microarrays, targeted gene sequencing, and clinical information: MSI, microsatellite stable (MSS)/Tumor Protein P53 (TP53)-inactive, MSS/TP53-active, and MSS/epithelial-mesenchymal transition (EMT) tumors. 43 MSS/EMT tumors have similar features to the GS subtype in TCGA classification, associated with diffuse-type histology, lowest rate of mutations, early-onset, highest risk of recurrence, development of peritoneal carcinomatosis, and worst prognosis among the four ACRG subtypes. In fact, the majority (80%) of MSS/EMT tumors included DGC. However, there are substantial differences between the GS subtype in TCGA and MSS/EMT subtype in ACRG. Although 57% of DGCs were classified into the GS subtype in the TCGA cohort, only 27% of DGCs exhibited MSS/EMT subtype in the ACRG cohort, with distribution across all ACRG subtypes. In addition, mutations of CHD1 and RHOA were less common in the MSS/EMT subtype than in the GS subtype.
Lei et al. identified three subtypes with distinct biological properties and chemotherapy sensitivities using a method of consensus hierarchical clustering with iterative feature selection: proliferative, metabolic, and mesenchymal tumors. 44 The mesenchymal subtype has features with cancer stem-like cells (CSCs) and is enriched in DGC. This subtype appears to have limited benefits from 5-fluorouracil (5-FU) chemotherapy. In a transcriptome analysis using the gene coexpression network, enriched mesenchymal stemness was a major driving force of DGC. 45
A growing number of studies have provided insight into the heterogeneity of DGC. A proteomic analysis showed three subtypes of DGC with distinct clinical outcomes based on the status of enriched immune response proteins and dysregulation in the cell cycle and EMT process. 46 A molecular signature of different prognostic subtypes of DGC was reported using RNA sequencing–based transcriptome data. 47 The intestinal-type-like subtype exhibited a signature associated with cell cycle or DNA repair, whereas the core diffuse-type subtype had poor prognosis and an enriched signature of EMT-associated functions, such as cell adhesion/migration and the TGF-β signaling pathway. In addition, DGC has a bimodal age distribution at diagnosis with early-onset and late-onset. Early-onset DGC has higher proportions of somatic mutations in CDH1 and TGF-β receptor 1 (TGFBR1) and lower proportions of RHOA mutations compared with late-onset DGC. 48
Collectively, although there is heterogeneity even among DGCs, most DGCs belong to the GS subtype in TCGA, MSS/EMT subtype in ACRG, and the mesenchymal subtype in Lei’s classification.
Aberrant signaling pathway in diffuse-type gastric cancer
Typically, IGC develops in the background of atrophic gastritis and subsequent intestinal metaplasia through a sequence of complicated multistep carcinogenesis. 49 Although the mechanism of developing DGC has not yet been fully understood, DGC may develop through a shorter, unidentified sequence of events from gastric epithelial cells. 50
A growing number of comprehensive genomic analyses of GC have shown that DGC exhibits different genetic and molecular profiles than IGC (Table 2 and Figure 1). In addition to mutations of genes associated with cell adhesion, cytoskeleton, and cell motility, including CDH1, RHOA, and Rho-associated coiled-coil-containing protein kinase (ROCK),18–21,51 multiplex profiling of peritoneal metastases from GC showed that DGC had high rates of heterozygous deletion of chromosome 3p, which encompasses multiple tumor suppressor genes, and duplication of 20q, which encompasses oncogenes. 40 Furthermore, DGC has frequent dysregulation of signaling pathways associated with hallmarks of cancer, including G2/M cell cycle checkpoint, mitotic spindle assembly, MYC, and inflammatory response. In a comparative study between signet ring cell phenotype and nonsignet ring cell phenotype among DGCs, the nonsignet ring cell phenotype had more frequent mutations of TP53, BRAF, phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha (PIK3CA), SMAD Family Member 4 (SMAD4), and RHOA, supporting heterogeneity of DGC. 30

Pivotal signaling pathways, including CLDN18.2, TGF-β, FGFR2, and MET, in DGC.
Molecular features according to the Lauren classification.

Amp, amplification; CDH1, cadherin 1; dMMR, deficient mismatch repair; EBV, Epstein–Barr virus; EGFR, endothelial growth factor receptor; FGFR2, fibroblast growing factors receptor-2; HER3, human epidermal growth factor receptor 3; LoEx: low expression; Meth: methylation; MSI-H, microsatellite instability-high; Mt: mutation; mTOR, mammalian target of the rapamycin; OverExp: overexpression; PD-1, programmed death-1; P-Exp: phospho-expression; VEGF, vascular endothelial growth factor.
The varied percentage of the PD-L1 positive expression was due to the different definitions of PD-L1 positivity and the cutoff values.
CDH1 is a tumor suppressor gene that codes for E-cadherin, which is a calcium-dependent transmembrane glycoprotein that forms the adherens junction via the extracellular domains, connects to the cytoskeleton via the cytoplasmic domains, and mediates cellular signaling. 85 Loss of its function confers diffused cellular morphology, migratory and invasive abilities, metastasis, and the EMT process.85,86 In DGC, E-cadherin expression is predominantly downregulated via somatic mutations or promoter hypermethylation of CDH1,18,19,40 and the loss of its expression is associated with an increased risk of early invasion and metastasis. 87 CDH1 germline mutations have been identified in patients with hereditary DGC, 28 and abnormal expression of E-cadherin occurs even in early gastric carcinomas, 88 suggesting that loss of CDH1 function is acquired early in the pathogenesis of DGC.
RHOA mutations predominantly occur in DGC, ranging from 15% to 25%,18,25,74 and the mutant RHOA acts as a gain-of-function. 25 The GS subtype exhibits fusions of ARHGAP6/26, which regulates RhoA activity as GTPase-activating proteins.18,20,21 Approximately 30% of GS subtypes have aberrant alterations in the components of the Rho signaling pathway. Activated RhoA signaling pathway regulates the actin cytoskeleton, cell migration, cytokinesis, the cell cycle, resistance to anoikis, and CSC phenotype with invasive ability and chemoresistance.25,74,89,90 In a mouse model, RHOA Y42 C, the most common RHOA hotspot mutation in DGC, coupled with loss of CDH1, induces metastatic DGC via actin cytoskeletal rearrangements and activation of Yes-associated protein1 (YAP1), phosphoinositide 3-kinase (PI3 K)-AKT, and β-catenin signaling pathways. 90 In 288 GC patients who underwent curative surgery, increased RHOA activity was an independent prognostic factor of OS in DGC patients, but not in IGC patients. 89 Thus, the aberrant activity of the RHOA signaling pathway has been implicated in the development and aggressive phenotype of DGC.
MET is a member of the RTK family and plays a key role in tumor cell proliferation, migration, invasion, survival, angiogenesis, and metastasis. 91 In a subset of GC, the MET signaling pathway is activated via the overexpression and gene amplification of MET. 92 The positivity rate of MET overexpression varies from 6% to 39% of GC,93,67 and MET gene amplification has been found in 2% to 7% of GC,67,68,94,95 which confers an unfavorable prognosis. 96 MET amplification is predominantly enriched in DGC compared with IGC,67,68,96,97 and is observed in both primary GC tissues and their corresponding cancer cells from malignant ascites. Thus, MET-amplified GC cells may have intrinsic potential for peritoneal dissemination, which is a major metastatic pattern of DGC.38,39
TGF-β is a multifunctional cytokine associated with tumor progression. 98 Approximately half of the GS subtype showed activation of the TGF-β signaling pathway in the TCGA cohort. 99 DGC is characterized by a rich fibrous stroma 34 and exhibits a high stromal super module expression with elevated levels of the TGF-β pathway in a comprehensive genomic meta-analysis using a network-modeling approach. 26 TGF-β ligands secreted by tumor cells alter normal fibroblasts to a myofibroblast-like phenotype, promoting tumor growth, vascularization, and metastasis. 100 Furthermore, activation of the TGF-β pathway has been observed in cancer-associated fibroblasts (CAFs) isolated from human DGCs, and the highly motile CAFs induced DGC with invasive abilities into the extracellular matrix and lymphatic vessels. 101 Thus, TGF-β signaling may act as a key indicator of a protumorigenic stromal environment in DGC. 98
Biological features of diffuse-type gastric cancer
DGC is often classified into mesenchymal subtype with features of CSC in Lei’s classification, 44 MSS/EMT subtype in ACRG cohort, 43 and GS subtype characterized by activation of RHOA, TGF-β, and Wnt pathways in TCGA classification,18,99 which have been implicated in the induction and maintenance of EMT and CSC phenotypes.89,102,103 These molecular subtypes support the association of DGC with EMT and CSC phenotypes. EMT is a cellular process of redifferentiation of epithelial cells into mesenchymal ones, which is implicated in embryogenesis, wound healing, carcinogenesis, and cancer progression. 103 In a clustering analysis of the TCGA cohort using an EMT gene set, the cluster with active EMT had an enriched TGF-β signaling pathway and MET amplification, which consisted of a high proportion (59%) of DGCs. 104 Importantly, EMT programs facilitate gastric CSC generation and expansion.105,106 CSCs are a relatively rare population of cancer cells that contribute to the driving force of tumorigenesis and metastasis due to their cancer stemness properties, including sphere formation, self-renewal, invasion, differentiation, antiapoptosis, immune evasion, and drug resistance.102,107,108 Thus, aggressive phenotypes of DGC, including chemoresistance, invasive and metastatic abilities, and poor survival outcomes, may be partially explained by the biological features of EMT and CSC phenotypes.
YAP1 is a downstream transcription coactivator of the Hippo signaling pathway, which acts not only as a prominent molecule for tumorigenesis, but also as an oncogenic driver in GC.109–113 YAP1 plays a crucial role in CSC expansion and properties in various tumor types, including GC.107,114,115 In fact, genetic knockdown of YAP1 has been found to suppress gastric CSC traits, tumorigenesis, and peritoneal metastasis in vitro and in vivo. 116 The Hippo pathway was also activated in the cluster with enriched EMT-related signatures in a comprehensive multiomic analysis of malignant ascites samples and their corresponding GC cell lines. 104 Thus, YAP1 is a promising target for gastric CSCs.
Treatment of patients with diffuse-type gastric cancer
Systemic chemotherapy includes three types of treatment: (1) cytotoxic agents, including fluoropyrimidines, platinum compounds, taxanes, trifluridine/tipiracil, and irinotecan; (2) immune checkpoint inhibitors (ICIs) targeting PD-1; and (3) molecular-targeted agents for HER2 or VEGFR2. Although there are different clinicopathological and molecular features in DGC and IGC, the same approach of treatment with systemic chemotherapy is used, regardless of histology types. In this section, the clinical efficacy of individual chemotherapy agents is described according to histology types.
Cytotoxic chemotherapy
The impact of cytotoxic chemotherapy on DGC has been evaluated in a few clinical trials and several exploratory analyses (Table 3). Generally, DGC is associated with low responsiveness to cytotoxic chemotherapy and chemoradiation, but the mechanisms of resistance are not yet understood. The adenosine triphosphate (ATP)-binding cassette (ABC) transporters confer multidrug resistance to tumor cells through an increased efflux of chemotherapy agents, and overexpression of ABC transporters has been reported predominantly in CSCs. 117 In a large-scale meta-analysis of GC transcriptome data, the negative association between stroma and cell proliferation in DGC relevant to a rich fibrous stroma may contribute to resistance to chemotherapy targeting active dividing cells. In addition, a high proportion of intratumoral stroma may directly inhibit the effects of chemotherapy by reducing drug delivery to tumor cells 118 and protecting cells against chemotherapy-induced apoptosis. 119
Results of phase III trials of cytotoxic agents according to histology types.

Adj, adjuvant chemotherapy; BSC, best supportive care; CAPOX, capecitabine + oxaliplatin; CI, confidence interval; DCS, docetaxel + cisplatin + S-1; DGC, diffuse-type gastric cancer; DOS, docetaxel + oxaliplatin + S-1; DTX, docetaxel; ECF, epirubicin + cisplatin + fluorouracil; FLOT, fluorouracil + leucovorin + oxaliplatin + docetaxel; FP, 5-fluorouracil + cisplatin; HR, hazard ratio; IGC, intestinal-type gastric cancer; N/A: not assessment; OS, overall survival; Peri, perioperative chemotherapy; Pre, preoperative chemotherapy; SOX, S-1 + oxaliplatin; SP (CS), S-1 + cisplatin; TAS102, Trifluridine/tipiracil; XP, capecitabine + cisplatin.
Disease-free survival (DFS).
Palliative chemotherapy
Combination of fluoropyrimidine with platinum is a standard first-line backbone regimen in GC, regardless of histology type.3,4 In real-world data, including 1303 patients who received chemotherapy, from the AGAMENON national registry research group, DGC patients showed decreased overall response rate (ORR) and increased mortality. 133 In a meta-analysis of 33 studies comprising 10,246 patients (4888 patients for IGC and 5358 patients for DGC) treated with chemotherapy, chemotherapy showed significantly improved OS in IGC patients compared with DGC patients (HR, 0.76; 95% CI, 0.71–0.82; p < 0.001). 17 Similar results were observed, even when subgroup analyses were performed according to the regimen of first-line chemotherapy. Among platinum compounds, oxaliplatin-based chemotherapy may be more efficacious than cisplatin-based chemotherapy for DGC patients.130,131
In a second-line setting, including 2311 patients in the AGAMENON group, DGC was also more refractory to chemotherapy than IGC. 134 In a salvage-line setting, a phase III TAGS trial evaluated the efficacy of perioperative trifluridine/tipiracil versus the best supportive care in 507 patients with refractory GC treatment and demonstrated more improved survival outcomes in IGC compared with DGC. 135 Collectively, DGC is associated with a poor prognosis and less sensitivity to chemotherapy.
Adjuvant/perioperative chemotherapy
In a large cohort study of 1290 locally advanced GC patients who underwent either primary surgery or preoperative chemotherapy followed by surgery, preoperative chemotherapy resulted in a deterioration of survival outcomes compared with primary surgery in DGC patients, in contrast to IGC patients, who had improved survival rates with preoperative chemotherapy. 136 Similarly, another large cohort study that assessed the survival impact of perioperative chemotherapy versus primary surgery in 924 patients with signet ring cell GC showed the detrimental effect of perioperative chemotherapy. 137 Thus, inherent chemoresistance may cause DGC patients to miss a chance of curative resection by delaying surgery through preoperative chemotherapy, especially in patients with signet ring cell GC. A phase II/III FREGAT trial (PRODIGE-19-FFCD1103-ADCI002), which compared perioperative chemotherapy (epirubicin, cisplatin, and 5-FU) with primary surgery followed by adjuvant chemotherapy, was conducted to elucidate whether primary surgery was a potential option for patients with resectable signet ring cell GC. 138 In phase II, the 2-year OS rates as a primary endpoint were met (60% in primary surgery and 54% in perioperative chemotherapy), and curative resection rates were 88% and 78%, respectively. 139 Currently, phase III is ongoing.
Immune therapy
Antitumor immune escape is often promoted by inhibitory immune checkpoint molecules, such as PD-1 and its ligand 1 (PD-L1), during the cancer-immunity cycle process. 140 PD-1 receptors on T cells bind to PD-L1, and the activated PD-1/PD-L1 signaling axis induces immune tolerance in the tumor microenvironment by disrupting the functioning of both cytotoxic and effector T cells.141,142 Currently, ICIs targeting PD-1/PD-L1 have dramatically changed therapeutic paradigms because of the durable clinical response in GC. However, the exploratory analyses of clinical trials suggest a poor response to treatment with ICIs in DGC (Table 4).
Results of phase II/III trials of molecular-targeted agents according to histology types.

Bema, Bemarituzumablumab; Bev, Bevacizumab; CAPOX, capecitabine + oxaliplatin; Cetu, Cetuximab; CI, confidence interval; CPT-11, irinotecan; DGC, diffuse-type gastric cancer; DTX, docetaxel; EGFR, endothelial growth factor receptor; EOX, epirubicin + oxaliplatin + capecitabine; Evero, Everolimus; FGFR2, fibroblast growing factors receptor-2; FOLFOX, fluorouracil + leucovorin + oxaliplatin; FP, 5-fluorouracil + cisplatin; HER2, human epidermal growth factor receptor 2; HR, hazard ratio; IGC, intestinal-type gastric cancer; Lap, Lapatinib; mTOR, mammalian target of the rapamycin; N/A, not assessment; Nivo, Nivolumab; Olap, Olaparib; Onar, Onartuzumab; OS, overall survival; PARP, poly ADP-ribose polymerase; PD-1, programmed death-1; Pembro, Pembrolizumab; Pertu, Pertuzumab; PTX, paclitaxel; Ram, Ramucirumab; Rilo, Rilotumumab; SOX, S-1 + oxaliplatin; Tmab, Trastuzumab; TDM-1, Trastuzumab emtansine; T-DXd, Trastuzumab deruxtecan; VEGFA, vascular endothelial growth factor A; VEGFR2, vascular endothelial growth factor receptor 2; XP, Capecitabine + cisplatin; Zolbe, Zolbetuximab.
Analysis of population of signet ring cell carcinoma.
From the antitumor immunogenic perspective, according to the molecular subtypes, DGC has several negative factors of sensitivity to ICIs. GS tumors exhibit lower PD-L1 expression, a lower tumor mutation burden that confers production of immunogenic neoantigens, and a lesser degree of immune cell signaling pathway as a tumor immunogenicity compared with MSI and EBV tumors, 18 leading to antitumor immune tolerance and evasion. In fact, DGC exhibits frequent heterozygous deletion of chromosome 9p24 involving PD-L1, 40 and consequently, low frequency of PD-L1 expression.157–159 Expression of major histocompatibility complex (MHC) class II is also low in DGC, which impairs antigen presentation. 160 In addition, DGC is associated with a mesenchymal-like phenotype,43–45 resulting in immune exhaustion due to the high expression of immune checkpoint T-cell immunoglobulin mucin receptor 3 (TIM3), its ligand galectin-9, another immune checkpoint V-domain Ig suppressor of T-cell activation (VISTA), and TGF-β1. 40 In the tumor microenvironment in DGC, there are low levels of tumor-infiltrating lymphocytes (TILs) 159 and frequent dysfunction of intratumoral CD8+ T cells. 161 Recently, multiplex profiling of the immune and stromal cell composition from peritoneal metastasis tissues has shown a distinct tumor microenvironment with lower levels of cytotoxic lymphocytes, monocytes, natural killer (NK) cells, and myeloid dendritic cells in DGC compared with IGC. 40 As other key components of the tumor microenvironment in DGC, abundant M2 tumor-associated macrophages (TAMs) play a pivotal role in promoting immunosuppressive signals.46,162 Collectively, DGC may be a ‘cold tumor’ with low TILs and low PD-L1 expression, 142 and the immunologically ignorant phenotype may have a poor response to ICIs. Therefore, novel treatment strategies to turn immunologically ‘cold’ tumors with poor immune activation into ‘hot’ tumors with strong immune infiltration are needed.
Molecular-targeted therapy
Based on the comprehensive molecular profiling of GC, many molecular-targeted agents have been developed. Unfortunately, most have failed to demonstrate treatment efficacy regardless of histology type in clinical trials (Table 4). Currently, clinically validated targeted molecules are HER2 and VEGFR2 in GC.
HER2 monoclonal antibody trastuzumab, in combination with chemotherapy, is now a preferred first-line treatment regimen for patients with HER2-positive GC.3,4 However, in a phase III ToGA trial, the survival benefit appeared to be limited in DGC patients (HR, 1.07; 95% CI, 0.56–2.05) in contrast to IGC patients (HR, 0.69; 95% CI, 0.54–0.88). 11 Recently, trastuzumab deruxtecan, an antibody-drug conjugate consisting of an anti-HER2 and a cytotoxic topoisomerase I inhibitor, showed remarkable improvement in ORR and OS compared with the physician’s choice of standard chemotherapy in a phase II DESTINY-Gastric01 trial. 12 A greater benefit for OS was found in DGC (HR, 0.38; 95% CI, 0.17–0.86) than in IGC (HR, 0.65; 95% CI, 0.39–1.07), along with that of ORR in DGC (66.7% versus 0%) compared with IGC (47.8% versus 21.6%). Trastuzuamb deruxtecan may be effective even for DGC, which is characterized by a scattered growth pattern 5 and a high incidence of intratumoral HER2 heterogeneity, 163 via a potent bystander effect due to a highly membrane-permeable payload. 164 However, the rate of HER2 positivity is rare in DGC, and the positive rates were 6.1% and 31.8% in DGC and IGC, respectively, 52 suggesting the limited benefits of HER2-targeted therapy in DGC patients.
Angiogenesis contributes to the progression of gastric tumorigenesis and metastasis by providing nutrition, growth factors, and an oxygen supply. 165 VEGF and its receptor VEGFR are one of the molecules responsible for angiogenesis, and functional genomic analysis has shown notably high expression of angiogenesis-related genes in DGC. 45 However, the dependency on angiogenesis is likely to be higher in IGC than in DGC. 166 Anti-VEGFR2 monoclonal antibody ramucirumab is the first molecular-targeted agent with survival benefits as a monotherapy in GC, as demonstrated in a phase III REGARD trial of ramucirumab versus placebo as a second or latter line in 355 GC patients. 13 Although the OS benefit of ramucirumab was better in DGC patients than in IGC patients, the PFS benefit was similar between DGC (HR, 0.49; 95% CI, 0.32–0.75) and IGC patients (HR, 0.46; 95% CI, 0.27–0.78). In a phase III RAINBOW trial that established ramucirumab plus paclitaxel over paclitaxel alone as a standard second-line regimen, 14 the addition of ramucirumab showed less benefit in DGC than IGC in terms of PFS (HR, 0.70; 95% CI, 0.52–0.93 versus HR, 0.53; 95% CI, 0.41–0.69) and OS (HR, 0.86; 95% CI, 064–1.15 versus HR, 0.71; 95% CI, 0.53–0.93). Similar findings were found in a phase III RAINFALL trial, which assessed whether the addition of ramucirumab to first-line chemotherapy prolonged OS in 645 patients. 149 These preclinical and clinical data suggest a lower efficacy of ramucirumab in DGC patients compared with IGC patients.
MET gene amplification is associated with DGC, and several MET inhibitors have been investigated in clinical trials. Onartuzumab is a fully humanized, monovalent monoclonal antibody. A randomized phase III METGastric trial of onartuzumab versus placebo in combination with first-line chemotherapy in MET expression-positive GC patients, assessed by immunohistochemistry (IHC), was conducted. 146 Enrollment was stopped early due to a lack of efficacy, and the clinical benefit was not observed even in a subset with an MET intensity of 2+ /3+. However, DGC showed a favorable HR of OS (HR, 0.82) compared with that of IGC (HR, 1.23). Rilotumumab is a fully human immunoglobulin (Ig) G2 monoclonal antibody. In a phase III RILOMET-1 trial of first-line chemotherapy plus rilotumumab versus chemotherapy plus placebo in MET expression-positive GC patients, 94 the addition of rilotumumab in combination with chemotherapy showed no effective clinical outcomes. Both the HR of OS and PFS showed poor trends in DGC compared with IGC. The main issues are coamplification of other RTKs such as HER2 and EGFR in MET-amplified GC, and heterogeneous MET amplification status between primary and metastatic lesions, leading to possible treatment resistance to MET inhibitors. 167
Promising targetable molecules in diffuse-type gastric cancer
Recently, two phase II trials demonstrated promising results of anti-CLND18.2 antibody (zolbetuximab) in the FAST trial 22 and anti-FGFR2-IIIb antibody (bemarituzumab) in FIGHT 23 (Table 5). Thus, CLDN18.2 and FGFR2-IIIb will be relevant therapeutic targets for DGC patients. This section focuses on CLND18.2 and FGFR2 in GC from both basic and preclinical viewpoints (Figure 1).
Results of phase II FAST and FIGHT trials.
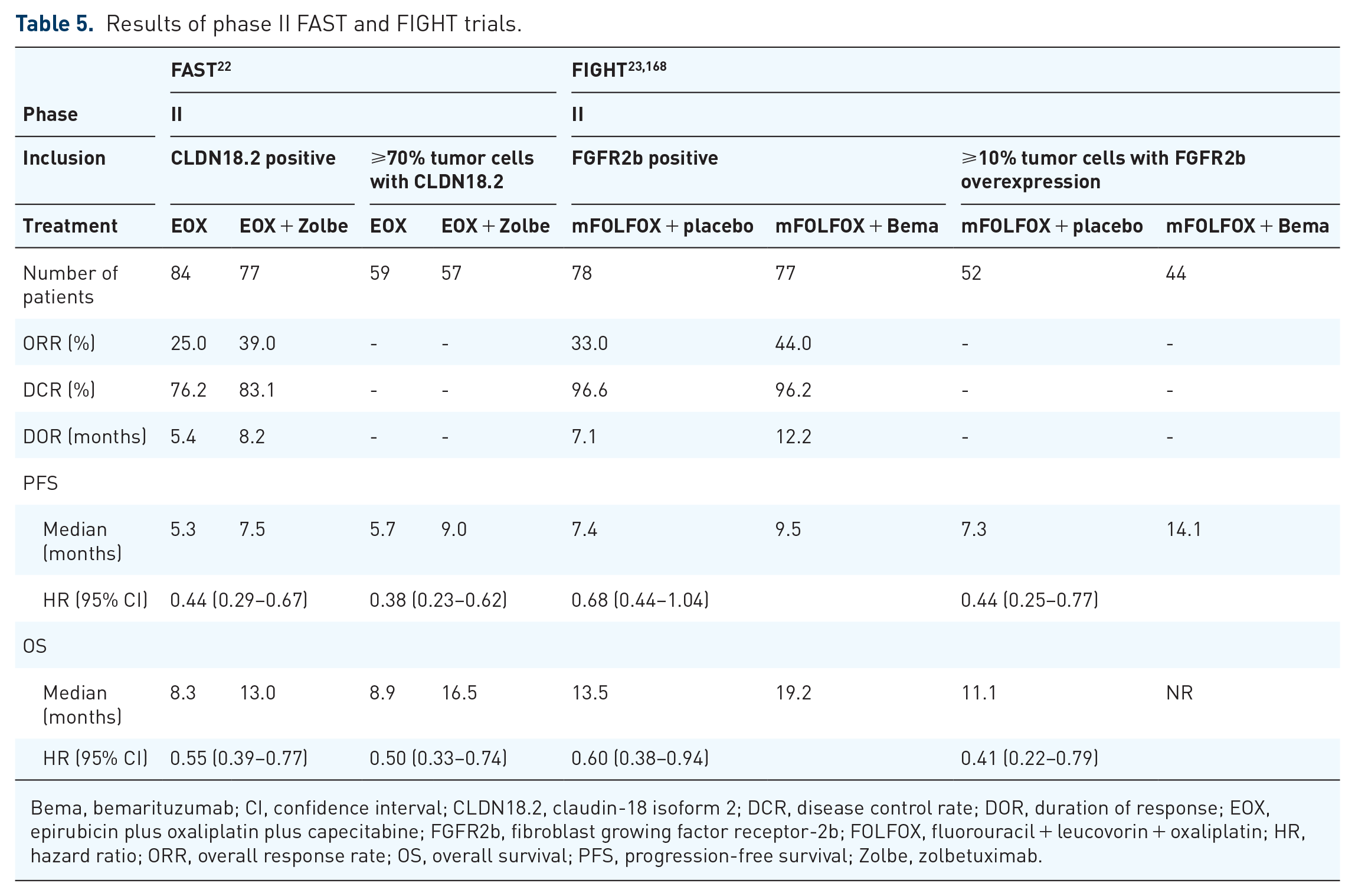
Bema, bemarituzumab; CI, confidence interval; CLDN18.2, claudin-18 isoform 2; DCR, disease control rate; DOR, duration of response; EOX, epirubicin plus oxaliplatin plus capecitabine; FGFR2b, fibroblast growing factor receptor-2b; FOLFOX, fluorouracil + leucovorin + oxaliplatin; HR, hazard ratio; ORR, overall response rate; OS, overall survival; PFS, progression-free survival; Zolbe, zolbetuximab.
Claudin 18.2 inhibitor
CLDNs are structural components of tight junction strands in the paracellular region that play critical roles in barrier function, permeability, paracellular transport, cell polarity, and signal transduction. 169 CLDN18 forms a paracellular barrier in the normal stomach, and its deficiency causes atrophic gastritis. 170 Two major CLDN18 isoforms, CLDN18.1 and CLDN18.2, are expressed almost exclusively in normal lungs and stomachs, respectively. 77 In the normal stomach, the expression of CLDN18.2 is strictly confined to differentiated epithelial cells in the gastric mucosa as a highly selective gastric lineage molecule. In GC, CLDN18.2 expression remains in primary and metastatic sites, and the high expression levels of CLDN18.2 (staining intensity of ⩾2+ in ⩾60% of tumor cells by IHC) were more frequently observed in DGC (75%) compared with IGC (46%). CLDN18-ARHGAP fusions are also predominantly detected in DGC. 20 In normal tissue, epitopes within the tight junction are generally inaccessible to intravenous antibodies. However, epitopes of CLDN18.2 may be exposed on the cell surface, possibly due to perturbations in cell polarity through malignant transformation and cell–cell detachment in DGC, leading to accessible drug targets. 77
Zolbetuximab (IMAB362) is a first-in-class chimeric IgG1 monoclonal anti-CLDN18.2 antibody. The two isoforms of CLDN18 are different in the N-terminal 69 amino acids, and the differences of amino acids in the first extracellular loop are only eight of the 51 amino acids. Nevertheless, zolbetuximab selectively binds to CLDN18.2, but not CLDN18.1, indicating lower cross-reactivity of zolbetuximab with CLDN18.1. In preclinical studies, treatment with zolbetuximab resulted in an antitumor effect by activating antibody-dependent cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) via immune effector stimulation. 171 Furthermore, zolbetuximab acts as a synergistic agent in combination with cytotoxic agents and enhances T-cell infiltration into the tumor microenvironment as an immunomodulator. 171
A phase II FAST trial was conducted to assess the clinical benefit of adding zolbetuximab to first-line epirubicin, oxaliplatin, and capecitabine (EOX) chemotherapy in recurrence or metastatic patients with CLDN18.2-positive GC, defined as moderate-to-strong CLDN18.2 expression in ⩾40% of tumor cells by IHC (Table 5).
22
A total of 334 (48.7%) of the 686 patients assessed by IHC had positive CLDN18.2 expression. Finally, 161 patients were randomly assigned to EOX (n = 84) and EOX plus zolbetuximab (n = 77). The primary endpoint, PFS, was met (HR, 0.44; 95% CI, 0.29–0.67; p < 0.001), and median OS as the secondary endpoint was 8.3 months for EOX and 13.0 months for EOX plus zolbetuximab (HR, 0.55; 95% CI, 0.39–0.77; p < 0.001). In terms of adverse events (AEs), nausea (81.8%) and vomiting (67.5%) were the most common, possibly due to the targeted effect on the normal stomach.
Considering the high frequency of CLDN18.2 overexpression in primary and metastatic tumors but its restricted expression in short-lived differentiated epithelial cells of gastric mucosa, 77 CLDN18.2 could be a promising molecule not only for a monoclonal antibody but also for an antigen targeted by antibody-drug conjugates (ADCs), bispecific T-cell engager (BiTE), and chimeric antigen receptor (CAR) T cells. ADCs comprise a cytotoxic payload conjugated by a linker to a monoclonal antibody against tumor-specific surface molecules, thereby enabling efficient drug delivery to tumor cells with minimum systemic exposure and off-target toxicity. 172 The redirection of T cells against tumors using CAR T cells or BiTEs has been demonstrated as a promising strategy for cancer treatment by activating T cells to kill tumor cells.173,174 Currently, several early phase trials of agents targeting CLDN18.2 are ongoing in CLDN18.2-positive tumors: (1) monoclonal antibodies including AB011 (NCT04400383), LM-102 (NCT05008445), TST001 (NCT04495296), MIL93 (NCT04671875), and NBL-015 (NCT05153096); (2) bispecific antibody targeting both CLDN18.2 and PD-L1 (Q-1802, NCT04856150); (3) anti-CLDN18.2 ADCs including CMG901 (NCT04805307), CPO102 (NCT05043987), SYSA1801 (NCT05009966), and LM-302 (NCT05161390); (4) BiTE targeting both CLDN18.2 and CD3 (AMG 910, NCT04260191); and (v) CAR T cells including CAR-CLDN18.2 T cells (NCT03874897), CT041 (NCT04404595), LY011 (NCT04966143), and LCAR-C18 S cells (NCT04467853).
FGFR2-IIIb inhibitor
The FGFR family consists of four highly conserved receptors (FGFR1, FGFR2, FGFR3, and FGFR4). The FGFR pathway is cancer-specifically dysregulated by their overexpression and genetic alterations in various tumor types, driving cancer development and progression.175,176 In GC, FGFR2 amplification is the most frequent genetic alteration among FGFR family members, ranging from 3% to 15% of GC, which confers an unfavorable prognosis.70,109,177–179 FGFF2 amplification is predominantly enriched in the GS molecular subtype in TCGA 18 or MSS/EMT subtype in ACRG, 43 indicating a key alteration in DGC.70,178 DGC also exhibits frequent FGFR2 overexpression with prognostic relevance, but IGC does not.70,180 FGFR2 isoforms (FGFR2-IIIb and IIIc) are determined by alternative splicing of a ternary extracellular immunoglobulin domain III, of which FGFR2-IIIb is predominantly overexpressed in GC, especially in DGC.70,180,181 In addition, FGFR ligands are overproduced in DGC cells and CAFs, leading to ligand-dependent FGFR2 activation through the paracrine and autocrine loops.182,183 Detailed information about the FGFR2 signaling pathway in GC has been reviewed elsewhere. 177 Preclinical studies have shown the antitumor efficacy of FGFR2 inhibitors in FGFR2-amplified DGC models. 184 Thus, the FGFR signaling pathway has attracted considerable attention as a targetable molecule, especially in DGC with FGFR2 aberrations.
A major area of drug development targeting FGFR is small molecule tyrosine kinase inhibitors (TKIs), and the therapeutic efficacy of FGFR-selective TKIs has been demonstrated for cholangiocarcinoma with FGFR2 fusions or rearrangements 185 and urothelial carcinoma with FGFR2/3 fusions or FGFR3 mutations. 186 Although several trials of FGFR-TKIs have been conducted in a subset of GC patients with FGFR aberrations, they have failed to demonstrate their clinical benefit.177,187 High-level clonal FGFR2 amplification may be an important predictive biomarker for selecting patients who would benefit from FGFR-TKIs. 188
Anti-FGFR2 monoclonal antibodies competitively bind to the extracellular domain of FGFR2 and block the activation of FGFR2 signaling, which has preclinical antitumor effects and less toxicity. 189 Bemarituzumab (FPA144) is a first-in-class humanized IgG1 monoclonal FGFR2-IIIb isoform-selective antibody glycoengineered for enhanced ADCC activity. 190 A randomized, double-blind, placebo-controlled phase II FIGHT trial was conducted to evaluate the clinical benefits of adding bemarituzumab to first-line modified oxaliplatin, 5-FU, and leucovorin (mFOLFOX6) chemotherapy in patients with FGFR2b-positive GC, defined as FGFR2-IIIb overexpression using IHC or FGFR2 gene amplification determined by circulating tumor DNA (ctDNA) (Table 5). 23 Of 910 patients who underwent prescreening, 275 (30.2%) were FGFR2b positive. Finally, 155 patients were randomly treated with bemarituzumab (n = 77) or a placebo (n = 78) in combination with mFOLFOX6. The primary endpoint, PFS, was met, with an improvement in median PFS of 9.5 months for bemarituzumab versus 7.4 months for the placebo (HR, 0.68; 95% CI, 0.44–1.04; p = .073). After an updated follow-up period of a median of 12.5 months, treatment with bemarituzumab resulted in prolonged OS, with a median OS of 19.2 months for bemarituzumab versus 13.5 months for placebo (HR, 0.60; 95% CI, 0.38–0.94). 168 The survival benefits increased with more homogeneous FGFR2-IIIb overexpression, with HRs of 0.44 for PFS (95% CI, 0.25–0.77) and 0.41 for OS (95% CI, 0.22–0.79) in GC patients with FGFR2-IIIb overexpression in ⩾10% of tumor cells. Stomatitis (31.6% versus 13.0%) and corneal AEs (67.1% versus 10.4%) were more common in bemarituzumab than the placebo. It remains unclear whether bemarituzumab has better treatment efficacy for patients with FGFR2-IIIb overexpressing DGC than IGC. However, considering the association of DGC with FGFR2-IIIb overexpression and FGFR2 amplification, bemarituzumab will be the main therapeutic pillar for patients with FGFR2-IIIb overexpressing DGC. A phase III study is warranted to confirm these results in a larger population of FGFR2b-positive GC.
Other potent molecular-targeted inhibitors
Although CDH1 is frequently mutated in DGC,18,19,40 it is not a conventional druggable molecule because of its function as a tumor suppressor gene and its loss of expression. Recently, synthetic lethality has been attracting attention as having the potential to target tumor cells that carry CDH1 mutations. The integrated genetic and drug screens, using breast tumor cells with CRISPR/Cas9-engineered CDH1 mutations or with homozygous deletion of CDH1, identified synthetic lethality between E-cadherin and the ROS proto-oncogene 1, receptor tyrosine kinase (ROS1).191,192 Because E-cadherin deficient tumor cells are dependent on ROS1, which is likely related to cytokinesis, ROS1 inhibitor crizotinib elicited synthetic lethality in the E-cadherin deficient GC cell models. 191 These preclinical findings provide the rationale for assessing the efficacy of ROS1 inhibitors. There is an ongoing phase II trial of crizotinib in E-cadherin negative DGC or CDH1 mutated solid tumors (NCT03620643).
The AT-rich interaction domain 1A (ARID1A), a subunit of the switch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complex, is one of the most commonly mutated genes across various tumor types. ARID1A mutations compromise diverse gene programs and cellular processes through the dysregulation of transcription, DNA repair, and chromatin segregation, thereby promoting tumorigenesis.193,194 In GC, ARID1A mutations are prevalent in the GS subtypes next to the EVB subtypes. 18 DGC are classified into early-onset and late-onset cancers, and ARID1A mutations were observed in 15–18% of early-onset DGC. 48 Several preclinical studies have reported crucial targets that induce synthetic lethality with ARID1A deficiency, such as enhancer of zeste homolog 2 (EZH2) histone methyltransferase that is a catalytic subunit of the polycomb repressive complex 2 (PRC2) that antagonizes the function of ARID1A, 195 BIRC5/Survivin that is a transcriptional regulatory module induced by ARID1A mutation, 194 and the glutamate-cysteine ligase synthetase catalytic subunit that is a rate-limiting enzyme for antioxidant glutathione synthesis. 196 Thus, the inhibition of molecules that create therapeutic vulnerability of the ARID1A-deficient tumor cells may be of clinical importance. In addition, ARID1A deficiency impairs the mismatch repair (MMR) system that plays a key role in correcting DNA replication errors, thereby resulting in an increased mutation burden, MSI-H genomic signature, TILs, and PD-L1 expression. 197 In xenograft models, ARID1A-deficient tumors, but not ARID1A-wild-type, were regressed by treatment with ICI. 197 GC patients with ARID1A mutation are likely to benefit from treatment with ICI.
Recurrent mutations of RHOA are a hallmark in DGC development.18,25,74,90 The genetic knockdown of RHOA repressed spheroid formation as a CSC phenotype and sensitized diffuse-type CSC cells to 5-FU and cisplatin chemotherapy. 89 RHOA inhibition also decreased the expression of EMT-related molecules. 198 In addition, ROCK is a downstream effector of RHOA, and treatment with the ROCK inhibitor (HA-1077) resulted in tumor regression in a transgenic GC mouse model. 199 As RHOA mutations promoted the activation and dependency of focal adhesion kinase (FAK), the small molecule inhibitors of FAK, including GSK-2256098, VS-6063, CEP-37440, VS-6062, VS-4718, and BI-853520, are considered to be a promising cancer therapy in RHOA mutant GC. 200 Furthermore, FAK may also serve as a target even for DGC without RHOA mutation. 90 These preclinical findings support the clinical utility of targeting the RHOA signaling pathway.
DGC are characterized by a rich fibrous stroma, which has been associated with overexpression in TGF-β signaling. 34 As TGF-β signaling contributes to tumor progression, metastasis, and drug resistance, 98 the TGF-β pathway has been pharmacologically targeted using monoclonal antibodies (SAR439459), small molecule inhibitors (vactosertib and galunisertib), ligand traps, and vaccines. 201 Based on a key immunosuppressive role of TGF-β signaling in the tumor microenvironment by restricting T-cell penetration in tumors, 202 a phase I trial of bintrapfusp alfa (M7824), a bifunctional fusion protein composed of a human anti-PD-L1 IgG1 monoclonal antibody fused with the extracellular domain of TGF-β receptor II, was conducted in patients with heavily pretreated GC. The ORR was 16%, and median duration of response was 8.7 months. 203 Furthermore, a rationale for dual blockade of TGF-β signaling and immune checkpoint molecules has been assessed in early phase trials using treatment with TGF-β inhibitor plus ICI (NCT03192345, NCT03724851), a bispecific antibody targeting both TGF-β and PD-L1 (Y101D, NCT05028556), and anti-CD73/TGF-β trap bifunctional antibody (GS-1423, NCT03954704).
Previous phase III trials have highlighted that biomarker selection for the specific molecular alteration is mandatory to enrich the efficacy of MET inhibitors.94,146 Savolitinib is a reversible MET-selective TKI, 204 and it has showed promising efficacy for MET-amplified GC patients assigned by targeted next-generation sequencing (NGS) using tissue DNA in a prospective biomarker-driven VIKTORY trial. 95 The ORR was 50% (10 in 20 GC patients) despite the second-line setting. Importantly, in the VIKTORY trial, the treatment efficacy was greater in GC patients with gene amplification detected by ctDNA than in tissue DNA. 95 Thus, the assessment of ctDNA is likely a strict approach to predicting treatment response to molecular-targeted agents by identifying high-level and clonal amplified tumors among GCs with intratumoral heterogeneity. A multicenter phase II trial to evaluate the efficacy and safety of savolitinib in MET-amplified GC patients is ongoing (NCT04923932).
Conclusion
DGC is associated with less chemosensitivity and an unfavorable prognosis through EMT and CSC phenotypes. There are no established therapeutic agents for DGC, so the development of novel treatment strategies for DGC is the most urgent need. The impressive results of two phase II trials demonstrate proof-of-concept, suggesting that anti-CLDN18.2 antibodies (zolbetuximab) and FGFR2-IIIb antibodies (bemarituzumab) are promising approaches for patients with CLDN18.2-positive and FGFR2b-positive GC, respectively. A new era of precision medicine for patients with DGC is dawning.
Footnotes
Author contributions
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: KY received speaker honoraria from Chugai Pharmaceutical Co. Ltd, Bristol-Myers Squibb, Merck Serono, Takeda, and Eli Lilly, and received consultant fee from Takeda Pharmaceutical Co. Ltd., and Honoraria from Tsumura Co. Ltd., Nihon Kayaku Co. Ltd., and Chugai Pharmaceutical Co. Ltd. AO received speaker honoraria from Merck Serono, Chugai, Takeda Pharmaceutical Co. Ltd., Daiichi Sankyo, and Ono Pharmaceutical.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.