
Abstract
Mortality from pancreatic ductal adenocarcinoma (PDAC) is increasing worldwide and effective new treatments are urgently needed. The current treatment of metastatic PDAC in fit patients is based on two chemotherapy combinations (FOLFIRINOX and gemcitabine plus nab-paclitaxel) which were validated more than 8 years ago. Although almost all treatments targeting specific molecular alterations have failed so far when administered to unselected patients, encouraging results were observed in the small subpopulations of patients with germline BRCA 1/2 mutations, and somatic gene fusions (neurotrophic tyrosine receptor kinase, Neuregulin 1, which are enriched in KRAS wild-type PDAC), KRAS G12C mutations, or microsatellite instability. While targeted tumor metabolism therapies and immunotherapy have been disappointing, they are still under investigation in combination with other drugs. Optimizing pharmacokinetics and adapting available chemotherapies based on molecular signatures are other promising avenues of research. This review evaluates the current expectations and limits of available treatments and analyses the existing trials. A permanent search for actionable vulnerabilities in PDAC tumor cells and microenvironments will probably result in a more personalized therapeutic approach, keeping in mind that supportive care must also play a major role if real clinical efficacy is to be achieved in these patients.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of a small category of cancers whose mortality is increasing and which will become the second leading cause of cancer death by 2030 in the United States and Europe. 1
In the early 2010s, the use of the 5-fluorouracil, irinotecan, and oxaliplatin (FOLFIRINOX), and gemcitabine plus nab-paclitaxel combinations improved the prognosis in selected patients.2,3 However, targeted therapies and immunotherapies, which have resulted in significant progress in patients with other tumor types, have so far failed in PDAC due to the biological characteristics of this entity.
Thus, the development of new, effective, and personalized treatments for PDAC is as urgent as ever. At the same time, there have been recent, encouraging results with the poly (ADP-ribose) polymerase (PARP) inhibitor, olaparib to treat PDAC in patients with BRCA1 or BRCA2 gene germline mutations (gBRCAm) 4 and in the development of various molecules targeting specific somatic genetic alterations.5–7 Nevertheless, the proportion of patients who can actually benefit from these treatments is still low either because these alterations are rare and can only be identified by technical means not available in all centers, or because validation to administer these treatments has not been obtained based on large randomized clinical trials.
This review discusses the hopes and limitations of recent advances in personalized treatment in patients with PDAC.
Therapeutic targets of interest for standard of care
HRD/BRCAness tumors and PARP inhibitors
Both BRCA1 and BRCA2 proteins are critical to the repair of double-strand DNA breaks by homologous recombination repair (HRR), a form of DNA repair using a homologous DNA sequence to guide this process at the double strand break (DSB; Table 1 and Figure 1). This conservative mechanism restores the original DNA sequence at the site of DNA damage (Lord and Ashworth). 8 Although targeted therapies have failed in PDAC in molecularly unselected patients, 9 progress has been made in recent years identifying small molecular subgroups of patients with targetable alterations. While the BRCA germline mutations are the most notable recent example, they are not alone. The concept of synthetic lethality was applied clinically using PARP inhibitors (PARPi) as single agent for maintenance therapy in patients with metastatic PDAC and the germline BRCA1 or BRCA2 mutation (gBRCAm) following platinum-based induction chemotherapy. For the first time, the POLO phase III trial confirmed the efficacy of a biomarker-driven approach in metastatic PDAC patients with the PARPi, olaparib. A significant benefit to progression-free survival (PFS) was demonstrated with maintenance olaparib versus placebo [median PFS: 7.4 months versus 3.8 months; hazard ratio (HR): 0.53; 95% confidence interval (CI): 0.35–0.82; p = 0.004]. 4
Progress and current limitations of medicine precision in PDAC treatment.

ATR, Rad3-related; BTK, bruton tyrosine kinase; CEA, carcinoembryonic antigen; CPI, checkpoint inhibitor; CTGF, connective tissue growth factor; ECM, extracellular matrix; EGFR, epidermal growth factor receptor; HA, hyaluronic acid; HLA, human leukocyte antigen; NTRK, neurotrophic tyrosine receptor kinase PAC, paclitaxel; PDAC, pancreatic ductal adenocarcinoma; PFS, progression-free survival; TMB, tumor mutational burden.
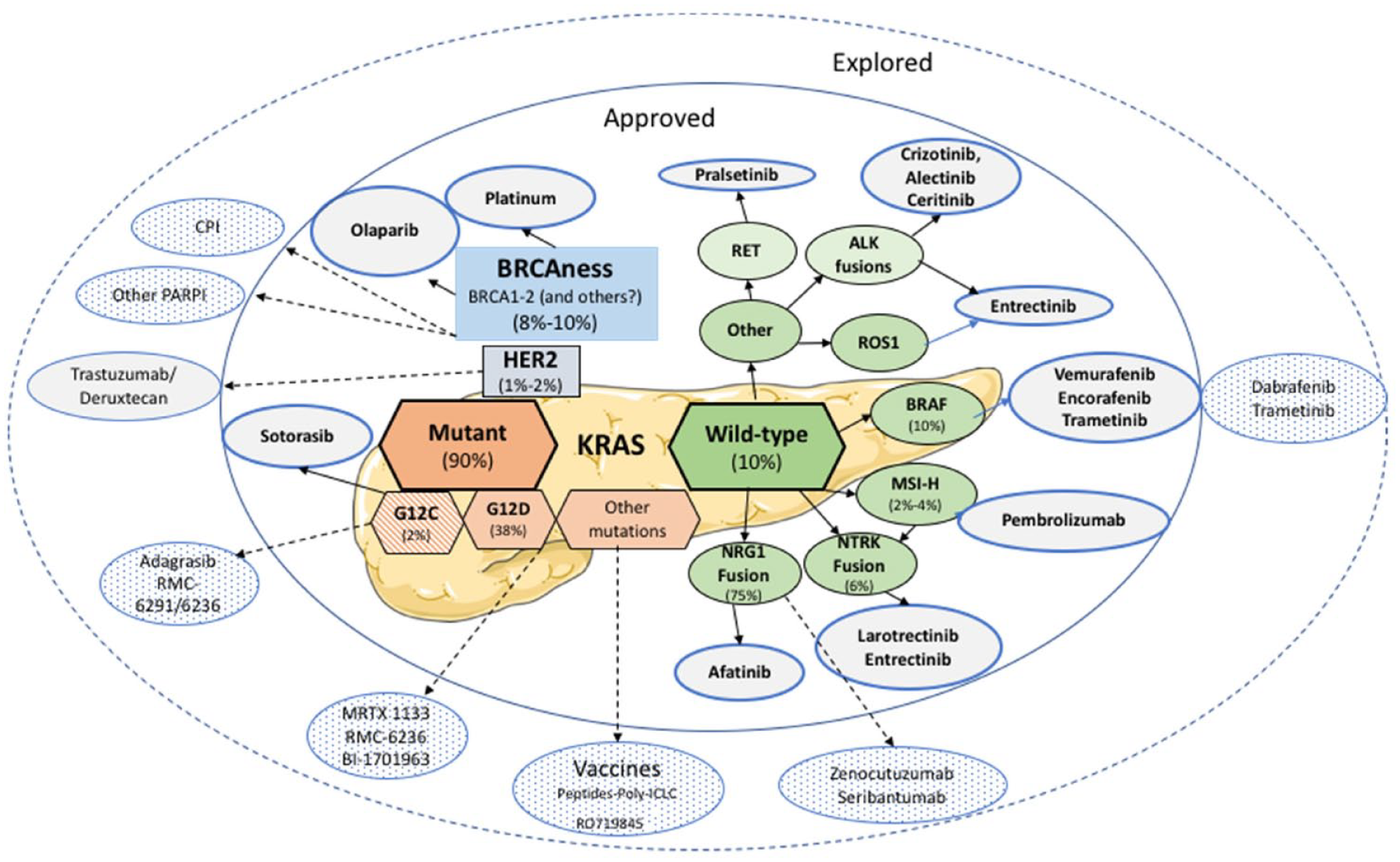
Potential active drugs (approved and explored) in PDAC according to actionable targets.
The impressive median overall survival (OS) in these patients with metastatic PDAC (19 months from the beginning of maintenance therapy) confirmed that gBRCAm confers sensitivity to both platinum-based chemotherapies and PARPi, resulting in favorable outcomes. In addition, Park et al. 32 later showed that the maximal therapeutic benefit with first-line platinum-based chemotherapy can be obtained in case of biallelic mutation in core HRR genes (BRCA1, BRCA2, and PALB2) compared to non-core genes (ATM, BAP1, BARD1, etc.). However, the POLO study did not show an advantage to OS in the olaparib arm compared to the placebo arm (median 19.0 versus 19.2 months; HR: 0.83; 95% CI: 0.56–1.22; p = 0.3487). 33 One explanation could be the extensive use of effective therapies following discontinuation of the study, resulting in tumor control in the placebo arm. This included reintroduction of platinum chemotherapy as well as the use of PARPi in 27.1% of patients, which was not allowed in the protocol. Despite this, the secondary endpoints (time to second disease progression or death, first and second subsequent cancer therapies or death and discontinuation), and estimated 3-year OS after randomization (33.9% versus 17.8%) were in favor of the olaparib arm. 33 Olaparib was approved by the Food and Drug Administration and European Medicines Agency at the end of 2019 for maintenance therapy in patients with germline BRCA1 or BRCA2 mutations (5–7% of PDAC patients, with BRCA2 representing about two-thirds of the gBRCAm) and metastatic PDAC, which has been controlled after at least 16 weeks of first-line platinum-based chemotherapy. 34 Systematic gBRCAm screening in PDAC patients is recommended in the National Comprehensive Cancer Network clinical guidelines. 35
Thus, for the first time, targeted therapy has been shown to be effective in the treatment of metastatic PDAC in a phase III trial, although these results have raised several questions that will be discussed below.
Moreover, in a randomized phase II trial, O’Reilly et al. 36 showed that there was no advantage to survival with a combination of upfront platinum-based chemotherapy and PARPi in gBRCAm PDAC patients. This suggests that patient selection based on tumor control during platinum chemotherapy can help identify patients who will benefit the most from PARPi. Indeed, about 20% of PDAC patients with gBRCAm are not platinum sensitive. 37
Resistance to PARPi
Approximately 40% of patients have a primary resistance to olaparib and a secondary resistance often occurs after several months of PARPi administration. There are numerous mechanisms of resistance to PARPi: (i) restoration of the HRR pathway through BRCA reversion mutations and/or epigenetic upregulation of the gene, (ii) loss of 53BP1 with rewiring of the non-homologous end-joint repair pathway, (iii) replication fork protection, (iv) upregulation of cellular drug efflux pumps, (v) reduction in PARP1 activity, or (vi) changes in the tumor microenvironment (TME).38,39 Integrating transcriptomic data with compound perturbation profiles could help identify drugs that can exploit induced vulnerability and thus overcome therapeutic resistance in multiple resistant clones. 40 Finally, another important approach would be detecting BRCA reversion mutations in circulating tumor DNA to anticipate the loss of PARPi efficacy. 41
Rad3-related (ATR), a key protein DNA for damage response, senses replication stress and induces signaling into the S and G2/M checkpoints to facilitate DNA break repair. Its activation is involved in resistance to PARPi. ATR inhibitors, such as VE-821, are a promising option to overcome these mechanisms of resistance to talazoparib and olaparib, mediated by inactivation of SLFN11 42 or to resensitize PARPi-resistant BRCA1-deficient cells which have restored HRR function to olaparib.43,44 Finally, epigenetic resensitization through DNA methyltransferase inhibition, cell cycle checkpoint inhibition, drug combinations with antiangiogenic agents, BET inhibition or G quadruplex stabilization or combining drugs upfront to prevent resistance, should also be explored.40,42
Efficacy of PARPi in non-core genes mutations?
There is very little information in the literature on the sensitivity to PARPi in tumors harboring a DNA damage repair mutation other than BRCA1 or BRCA2 (e.g. ATM, BAP1, BARD1, etc.) or on the activity of the different PARPi on somatic versus germline ‘BRCAness’ mutations.45,46 Somatic ‘BRCAness’ mutations could be present in up to 15–17% of PDAC patients. 47 The activity of PARPi has been reported to be heterogeneous in other cancers depending on the presence of a germline or somatic mutation, and the gene involved (e.g. in prostate or ovarian cancer).48,49 However, there is not enough existing data to extend the use of olaparib to germline mutations other than BRCA1 or BRCA2 or to somatic ‘BRCAness’ mutations, and prospective trials are needed. The MAZEPPA French trial (NCT04348045) is an open-label, phase II study to assess the efficacy of genomic-driven maintenance therapy for PFS in metastatic PDAC patients whose tumor has been controlled after 4 months of mFOLFIRINOX chemotherapy. In this trial, patients with a BRCAness somatic profile receive olaparib while those with no BRCAness profile and with a KRAS mutation are randomized to receive durvalumab plus selumetinib or FOLFIRI.
Which patients with PDAC should be tested for BRCAness mutations?
Ideally, all patients with PDAC should be tested 50 to identify whether a gBRCAm has immediate and individual theragnostic value. Moreover, diagnosis of a germline mutation can directly influence the patient’s relatives and requires an oncogenetic consultation with a longer delay. However, it should be remembered that the primary goal of gBRCAm testing is theragnostic because it was not broadly indicated before the POLO trial results, except in patients with a family history suggesting a potential genetic predisposition.
It has been shown that about 40–60% of patients with gBRCAm do not declare a family history of BRCA-related cancers (PDAC, breast, ovarian or prostate cancer) 51 either because they are not asked this question by the doctor, or the patient does not know or has forgotten the family history, or the family history is non-informative (incomplete penetrance). 52 Published data on referral rates in PDAC are limited. A US study reported that one-third of patients referred for genetic counseling did not complete germline testing for various reasons (worsening of disease severity, lack of follow-up, etc.). 53 An Italian real-life study also showed that germinal BRCA 1/2 pathogenic variants were found in 8.1% of the population, and 29% of patients had a family history of potentially BRCA-associated tumors (breast ovarian, prostate, or pancreas). However, in the same study, 45% of patients with a gBRCA mutation had no or unknown family history of BRCAness. 54
Thus, a family history of cancers from the BRCA-spectrum should not be considered necessary to select patients for gBRCAm screening. It would seem that the gastroenterology/oncology medical community needs to be more aware of the importance of collecting family information and patient screening at the diagnosis of PDAC. However, it may not be cost-effective to screen all PDAC patients. For example, the gBRCAm might only be searched for in patients who are potential olaparib candidates, that is, who are fit for platinum-based chemotherapy.
When should patients be tested for BRCAness mutations?
In US guidelines, early testing for BRCA mutations (excluding variants of unknown significance) in all PDAC patients is now recommended with both somatic and germline analyses. 50 Thus, testing can be proposed at diagnosis when planning for biologically adapted treatment (i.e. platinum-based chemotherapy and PARPi maintenance) whenever possible. In practice however, proposing this test during the first meeting with patients who are worried about this life-threatening disease can be difficult. Presenting testing as a potential opportunity to have access to additional therapeutic options can minimize a patient’s feeling of guilt for her/his relatives about transmitting the mutation to her/his children if a germline mutation is discovered. Another problem is the potential failure of upfront induction chemotherapy (17% of patients in the POLO study) which prevents the use of maintenance treatment such as PARPi. 4 Thus, it seems to be acceptable to wait until the first tumor evaluation to select patients for mBRCAg testing.
This explains why somatic testing was performed in the MAZEPPA trial after 2 months (four cycles) of mFOLFIRINOX to ensure tumor control. Finally, testing can encounter organizational difficulties to obtain the result within a sufficiently short period of time in relation to the theragnostic aim, or reimbursement depending on the country and center. Moreover, there is also the risk of poor quality material and thus to non-informative results with somatic testing.
Molecular alterations in KRAS wild-type PDAC
A hallmark of PDAC is the prevalence of the oncogenic mutation in the KRAS gene. 55 Activating KRAS mutations occur in approximately 90% of cases. They are an early and major event in pancreatic carcinogenesis. They result in permanent activation of the KRAS small GTPase protein, which acts as a molecular switch to activate various intracellular signaling pathways (including the MAP kinases and mTOR pathway) and transcription factors inducing cell proliferation, invasion, and survival.56–58 KRAS-driven tumors are largely refractory to anticancer therapies.
KRAS wild-type PDAC in 10% of PDACs, 59 an entity with a good prognosis in the literature, 60 offers therapeutic opportunities due to an enrichment in alternative, potentially actionable, oncogenic drivers which are (almost always) mutually exclusive with the KRAS mutation: (i) fusion genes and (ii) BRAF alterations. 58
In a study by Philip et al. 59 based on whole exome sequencing of 2483 PDACs, MSI-H/dMMR tumors were found in 4.7% of the cases versus 0.7%, tumor mutational burden (TMB)-high in 4.5% versus 1% and a larger infiltration of CD8+ T cells, natural killers, and dendritic cells (DCs) which could make these KRAS wild-type tumors more sensitive to immune checkpoint inhibitors (CPIs).
Windon et al. 61 reported that patients with KRAS wild-type PDACs have a longer median OS (KRAS wild-type, 957 days versus KRAS mutant, 531 days; p = 0.026) than those without. Similarly, Ogura et al. 62 found significantly better OS in patients with KRAS wild-type PDAC (479 versus 255 days, p = 0.03).
In addition, KRAS wild-type PDAC appears to be sensitive to epidermal growth factor receptor (EGFR) inhibition. 63 Accordingly, the NOTABLE phase III trial, evaluating gemcitabine plus nimotuzumab (a humanized anti-EGFR monoclonal antibody) combination versus gemcitabine plus placebo have shown good results in the experimental arm with a significant improvement of OS [10.9 versus 8.5 months, HR: 0.50 (0.06–0.94), p = 0.024] in KRAS wild-type advanced PDACs. 64
Previous studies examining the genomic profiles of KRAS wild-type PDAC have shown considerable heterogeneity, with GNAS, BRAF, and CTNNB1 alterations and additional RAS pathway genes identified as oncogenic drivers and kinase fusions in FGFR2, RAF, ALK, RET, MET, neurotrophic tyrosine receptor kinase 1 (NTRK1), ERBB4, and FGFR3.47,59,65 In particular, actionable somatic alterations were frequently identified in early onset pancreatic cancer, enriched in the RAS wild-type subgroup. For example, in a study performed at Sloan Kettering Memorial hospital in 132 early-onset PDAC, 15.9% had RAS wild-type PDAC with actionable alterations, including ETV6-NTRK3, TPR-NTRK1, SCLA5-Neuregulin 1 (NRG1), as well as ATP1B1-NRG1 fusions, IDH1 R132C mutations, or mismatch repair deficiencies. 66 Another whole genome and transcriptome sequencing study that was performed to identify clinically actionable genomic alterations in young adults with PDAC alterations identified recurrent NRG1 rearrangements that are supposed to drive PDAC development through aberrant ERBB receptor–mediated signaling. 67
NRG1 fusions
NRG1 is a ligand that binds to human epidermal growth factor receptor 3 (HER3), promoting HER2/HER3 heterodimerization and activation of PI3K/AKT/mTOR signaling. Chromosomal rearrangements involving NRG1 are rare oncogenic drivers in solid tumors, enriched in KRASwt PDAC and lung invasive mucinous adenocarcinoma.68,69 NRG1 fusion positive (NRG1+) in vitro and in vivo models are sensitive to HER2/HER3 directed therapy.63,70–73 There are over 20 partners of NRG1 fusions in solid tumors and some reports reveal common partners according to cancer type. CD74 has been shown to be the most frequent and ATP1B1, CDH1, and VTCN1 were detected as a partner of NRG1 fusion in PDAC.68,69.
In a study by Jones et al. 74 in 47 patients with PDAC who underwent comprehensive whole genome and transcriptome sequencing and analysis, two of the three patients with NRG1 fusion-positive tumors were treated with afatinib (tyrosine kinase inhibitor targeting HER2 and HER3) and had a rapid, significant response on therapy.
A recent phase II basket trial with zenocutuzumab (MCLA-128), a bispecific antibody targeting NRG1 fusion signaling, was launched in patients with NRG1 fusion-positive PDAC, non-small-cell lung cancer (NSCLC), and other solid tumors (NCT02912949). Schram et al. 75 reported a partial response in 42% of patients with MCLA-128 in a series of PDAC patients in this ongoing trial. Zenocutuzumab induced rapid and major radiological tumor regression and biomarker responses in heavily pretreated metastatic KRAS wild-type, NRG1-positive pancreatic cancer, with a good tolerance.
The rarity of NRG1 fusions and the diversity of fusion partners may make their detection challenging. It is important to note that DNA sequencing is not sufficiently sensitive to detect these fusions. Thus, RNA sequencing is considered to be the gold standard for this purpose, and offers the greatest likelihood of identifying NRG1 gene fusions when the identity of the fusion partner is unknown.76–78
NTRK fusion
The genes NTRK1, NTRK2, and NTRK3, respectively, encode tyrosine kinase receptors tropomyosin-related kinases A, B, and C (TRKA, TRKB, and TRKC). Fusion of a variety of different partners with NTRK1, NTRK2, or NTRK3 results in oncogenic proteins that act for constitutive activation of the RAS-MEK-extracellular signal-regulated kinase (ERK), diacylglycerol-inositol-1,4,5-trisphosphate, and PI3K-AKT signaling pathways.69,79
However, these NTRK fusions are uncommon (<0.5% of PDAC), and mainly found in rare pediatric tumors, although they can also be encountered in common digestive cancers such as colorectal cancer, especially in case of RAS/BRAF wild-type dMMR/MSI-H tumors (with an enrichment in up to 40% of tumors), or in aggressive cancers, such as PDAC.12,80,81 In a study by Allen et al. 82 , whole genome sequencing and RNAseq were performed in a series of 400 patients with resected or locally advanced/metastatic PDAC. The prevalence of NTRK fusions was 0.8% (3/400), but it was 6.25% (2/32) in KRAS wild-type tumors. Since the results of the NAVIGATE trial, these fusions represent a histoagnostic alteration that can be targeted. In this phase II clinical trial that explores the efficacy and long-term safety of larotrectinib, 121 (79%) patients with NTRK fusion-positive solid tumors had an objective tumor response. One of the two patients with PDAC were responders.83,84 In another study, two patients with PDAC and NTRK fusions achieved a partial response to entrectinib. 7
Other targetable fusions have also been described but are uncommon (each < 0.5%: RET, enriched in acinar pancreatic cancer, ALK, FGFR2, etc.).
KRAS mutations
Direct KRAS blockade remains a challenge, and successful inhibition of a key downstream effector pathway, the RAF-MEK-ERK cascade has been limited because of activation of feedback networks that keep the pathway in check (Table 2).85,86 Overall, the unsuccessful clinical trials to prevent localization of the RAS protein to the plasma membrane and inhibit downstream oncogenic signaling by targeting KRAS effectors in MEK1/2, ERK1/2, or AKT alone or in combination have led to the conclusion that KRAS was an ‘undruggable’ target. 73
Trials with drugs targeting KRAS mutations.

ERK, extracellular signal-regulated kinase; HLA, human leukocyte antigen; TCRs, T-cell receptors.
G12D (38%) and G12V (28%) are the most common mutations in PDAC.87–89 In recent years, inhibitors have been developed to target the KRASG12C mutation (adagrasib, sotorasib), based on the physicochemical particularities of the cysteine amino acid (G12C). The mutated cysteine resides next to a pocket (P2) of the switch II region. The P2 pocket is only present in the inactive GDP-bound conformation of KRAS and has been exploited to establish covalent inhibitors of KRASG12C.14,90,91 They bind to KRAS-GDP-bound and lock it in its ‘off’ form.90,92 The targeting of this mutation was initially developed in lung and colorectal cancers. Early clinical trials of these inhibitors were promising in the KRASG12C mutant PDAC, which is only found in 1–2% of PDAC, in early clinical trials.93–95
Preclinical studies showed that sotorasib (AMG510) inhibited nearly all detectable phosphorylation of ERK, a key downstream effector of KRAS, leading to durable complete tumor regression in mice bearing KRASG12C tumors. Preclinically, AMG510 selectively targeted KRASG12C tumors, caused durable regression as a monotherapy, and could be combined with cytotoxic and targeted agents to synergistically kill tumor cells. Moreover, AMG510 treatment led to an inflamed TME that was highly responsive to immune checkpoint inhibition. 96
In the phase I study by Hong et al. 93 in advanced solid tumors, no dose-limiting toxic effects were observed with sotorasib and no treatment-related adverse events resulted in death. Four patients with tumor types other than lung or colorectal cancers had a confirmed partial response, including one with PDAC. 93 In all, 12 patients with KRASG12C PDAC were enrolled in the next phase I/II study with sotorasib (NCT03600883). One out of 11 patients who could be evaluated had a partial response, 8 patients had stable disease, and 2 patients had progressive disease. 93 Recently, the combined data from the phase I/II CodeBreaK100 trial in 38 PDAC patients treated with sotorasib, the objective response rate (ORR) was 21% and disease control rate (DCR) 84.2% among patients who had already received at least one therapy for PDAC.94,97
Adagrasib (MRTX849) is another KRAS G12C inhibitor. At the ASCO GI 2022, Bekaii-Saab et al. 95 presented the updated results of the KRYSTAL-1 study (NCT03785249) about the activity and safety of adagrasib as monotherapy in 30 patients with previously treated, inoperable or metastatic KRASG12C-mutant GI tumors other than colorectal cancer. Among the 12 patients with PDAC in this series (median prior lines of therapy: n = 3; median follow-up 8.1 months), a partial response was obtained in 5 out of 10 patients who could be evaluated. All patients achieved a DCR and the PFS was 6.6 months (95% CI 1.0–9.7). Adverse events were diarrhea (43%), vomiting (43%), and fatigue (29%) (grade 3/4: 21%).
More effective combinations of various targeted therapies have been studied preclinically with KRAS G12C inhibitors in colorectal cancer (e.g. combination of adagrasib with anti-EGFR cetuximab in the KRYSTAL-1 study) and NSCLC.15,98 Multiple combinations are now in clinical trials (Table 2), such as new combination arms in the CodeBreak 101 trial combining sotorasib with a MEK inhibitor, PD-1 inhibitor, SHP2 inhibitor, pan-ERBB inhibitor, PD-L1 inhibitor, CDK inhibitor, or mTOR inhibitor (NCT04185883). The KRYSTAL-2 trial of adagrasib explores the role of the SHP2 inhibitor TNO155 (NCT04330664). Multiple additional promising G12C inhibitors are currently in clinical development as well, including JNJ-74699157 (NCT04006301), GDC-6036 (NCT04449874), and JDQ443 (NCT04699188) 99 in PDAC patients. Whether the depth and duration of response will be comparable to that obtained in lung cancer (up to 40% response rate) remains to be determined.
KRASG12D is a much more common alteration in PDAC and it represents a third of KRAS mutations. As shown in recent attempts, it lacks a reactive residue adjacent to the switch II pocket16,100 and thus new selected drugs must be designed. The G12D inhibitor MRTX1133 was discovered through an intensive structure-based search during adagrasib synthesis and high-resolution X-ray crystal structures. Its efficacy has been found to be promising in mutant Panc 03-04 xenograft mouse tumor models, 16 which is important because the KRAS missense mutation G12D is the most predominant variant in human malignancies (35%), and 39% of PDAC patients harbor this mutation (Table 2).
Other inhibitors, such as SOS1:KRAS interaction inhibitors, could be effective on a larger spectrum of KRAS mutations. BI-3406, a potent and selective SOS1:KRAS interaction inhibitor, has been shown to decrease the formation of GTP-loaded RAS and reduce cell proliferation of a large fraction of KRASG12C and non-G12C-driven cancers in vitro and in vivo. In mouse models, BI-3406 attenuates feedback reactivation by MEK inhibitors and enhances sensitivity of KRAS-dependent cancers to MEK inhibition, resulting in tumor regression at well-tolerated doses. 17
Mutant KRAS drives immune evasion in cancer cells by the upregulation of inhibitory immune checkpoint expression, the downregulation of MHC-I expression on tumor cells, the enhancement of cytokines and chemokines, and the recruitment of suppressive immune cells. 19 Mutant KRAS neoantigens presented on human HLA and T cells can recognize mutant KRAS cells and paved the way of strategies for targeting KRAS immunologically with vaccines alone or in combination. 101 A highly promising approach using T-cell engineering targeting the KRAS G12D hotspot mutation will be described further on in the chimeric antigen receptor (CAR)-T cells in section “CAR-T-cell development.” 102
Targetable BRAF mutations
BRAF mutations, which are generally mutually exclusive with KRAS, are present in 3% of PDAC but up to 10% of KRAS wild-type PDAC. 103 V600E BRAF mutations are prominent. Other alterations, such as non-V600E BRAF in-frame insertions or deletions or gene fusions, have been reported in 1% of patients with PDAC. 10
A combination of the BRAFV600E inhibitor vemurafenib and the MEK inhibitor cobimetinib resulted in an objective tumor response in a patient with poorly differentiated, V600E mutant PDAC. 104 In another report, six patients, five with a V600E mutation and one with CUX1-BRAF fusion, received vemurafenib single agent. 105 In a series of the ‘Know Your Tumor’ program, two of 18 PDAC patients with BRAF-mutant PDAC received the dabrafenib/trametinib (BRAF/MEK-inhibitor) combination: one patient with tumor V600E mutation had a sustained response, while treatment failed in the other with KRASG12A/BRAF K601N. 106
Immunotherapy in PDAC: microsatellite instability and high tumor mutational burden
The resistance of PDAC to immunotherapies could be due in part to a paucity of neoantigen-reactive tumor-infiltrating lymphocytes, which can mainly be explained by the low immunogenicity and non-inflamed phenotype of PDAC. The abundant stroma generates a hypoxic microenvironment and drives the recruitment of immunosuppressive cells through cancer-associated-fibroblast activation and transforming growth factor β (TGFβ) secretion. 107 Nevertheless, we have begun to identify subgroups of responders to immunotherapies.
DMMR/MSI-H tumors
The deficient deficient mismatch repair (dMMR)/ microsatellite instability-high (MSI-H) phenotype could be an interesting target even in the presence of sporadic or Lynch syndrome (LS) tumors.108,109 In a multicenter study of immunohistochemical analyses of 445 PDAC specimens, 1.6% of tumor samples were dMMR/MSI-H. 110 This rate was 2.61% in a meta-analysis of 34 studies pooling 8323 PDAC patients. 60 However, the dMMR/MSI-H PDAC rate in the latter study may have been overestimated, since some studies focused on PDAC subtypes enriched by this molecular alteration. dMMR/MSI-H in PDAC is strongly associated with the medullary and mucinous/colloid histological phenotype. This is associated with a KRAS/TP53 wild-type molecular background and a higher rate of JAK (JAK1 and JAK3) and KMT2 (KMT2C and KMT2D) gene mutations, with no benefit to survival, like in colorectal cancer. The genomes of dMMR/MSI-H tumors harbor hundreds to thousands of somatic mutations that encode potential neoantigens. Thus, often immunogenic, triggering upregulation of immune checkpoint proteins. In the phase II KEYNOTE-158 study testing the anti-programmed death-1 therapy pembrolizumab in pretreated patients with advanced non-colorectal dMMR/MSI-H tumors, the primary endpoint was the ORR according to Response Evaluation Criteria in Solid Tumors version 1.1, as assessed by an independent central radiologic review. 111 While there was a clear clinical benefit using this single immunotherapy agent, the ORR in the subgroup of patients with PDAC was only 18.2%, which is low compared to endometrial (57.1%), gastric (45.8%), cholangiocarcinoma (40.9%), small intestine (42.1%), or ovarian (33.3%) cancers. 111 This could be related to the specific unfavorable immune microenvironment or the misdiagnosis of PDAC or MSI-H. 11
dMMR/MSI-H PDACs has been shown to have a higher density of CD8+ T cells at the invasive front (p = 0.08) and expression of the CD274 molecule more often than other PDACs (p = 0.05). 110 Nearly 10% of PDAC patients harbor germline variants, although most lack somatic second hits and its therapeutic significance has not been defined. 112 In a multicenter study of 289 patients with resected PDACs who were not preselected for age or personal/family cancer history, targeted germline analysis of 24 genes related to an inherited cancer predisposition identified pathogenic/probably pathogenic germline variants in nearly 10% of patients, including 7.3% with variants in genes related to dMMR genes. Patients with a LS have an 8.6-fold greater risk of developing PDAC than the general population 113 with a cumulated risk of PDAC of 3.7% and tumors exhibiting a medullary appearance with prominent lymphocytic infiltration. 114
The literature on somatic-only dMMR status is more scarce. hMLH1 promoter methylation was detected in 60.0% of a series of 90 operated patients. 115 Finally, dMMR/MSI-H is often a genetic alteration enriched in KRAS wild-type PDAC. 60 These observations are the reason for the recent guidelines for dMMR somatic testing in PDAC and universal germline testing of dMMR tumors.35,116
High tumor mutational burden
Except for MSI tumors, immunotherapy has thus far failed in PDAC, even with ICI combinations 117 or chemotherapy. 118 TMB in PDAC is about one mutation per megabase (mut/Mb), which is 10-fold less than that of melanoma or lung carcinoma. In addition, PDACs are characterized by low T-cell recruitment and dense desmoplastic stroma resulting in high interstitial pressure, poor tumor perfusion, and hypoxia. Activation of fibroblasts then releases cytokines [TGFβ, interleukin (IL)-6, colony- stimulating factor 1 (CSF1) etc.] leading to immunosuppressive cell recruitment (M2 macrophages, regulatory T cells (TREG), myeloid derived suppressor cells (MDSC), as well as exclusion and effector T-cell anergy, which may explain the failure of immune CPI treatments. 107
Response rates with pembrolizumab have been shown to be significant on tissue TMB (tTMB) data The rate in the KEYNOTE-158 study was approximately 30% for tTMB-high tumors (at least 10 mutations per megabase) (Table 1). 20 This response rate could mainly be due to the potential of tumor mutations to generate immunogenic neoantigens. There was no benefit to OS in the recent PA.7 randomized phase II trial evaluating gemcitabine-nab-paclitaxel with durvalumab and tremelimumab. However, tTMB and ATM were identified as predictive biomarkers of response. 118 Nevertheless, results are contradictory. In another study, data from over 10,000 tumors from patients included in The Cancer Genome Atlas were analyzed to evaluate the association of t-TMB with CPI treatment outcomes. This analysis did not support the use of TMB-H as a biomarker for treatment outcomes of several cancer types with CPI. 119 Further studies are needed to evaluate other tumor-specific types especially in PDAC for which data are lacking.20,119
Immunotherapy proficient MMR tumors
Except for dMMR/MSI-H PDAC, single-agent immunotherapy has not been found to be effective in PDAC because this entity combines poor tumor immunogenicity with a low mutation rate (Table 3). 107 Nevertheless, immunotherapy agents are being tested in numerous combinations with various molecules including tyrosine kinase inhibitors or classic gemcitabine-nab-paclitaxel (NCT04498689, NCT04181645, and NCT03989310) or FOLFIRINOX schemas (NCT04324307, NCT03977272, and NCT04181645) (Table 3). It is interesting to note that T-cell reactivity to high-quality tumor neoantigens has been shown to be a characteristic in long-term survivors thus could lead to reposition immunotherapy in a subset of patients with MMR PDAC. To illustrate this, a promising approach using T-cell engineering will be described further on in this review (cf section “CAR-T-cell development").
Combination of immunotherapy with other drugs in PDAC patients: trials in progress.
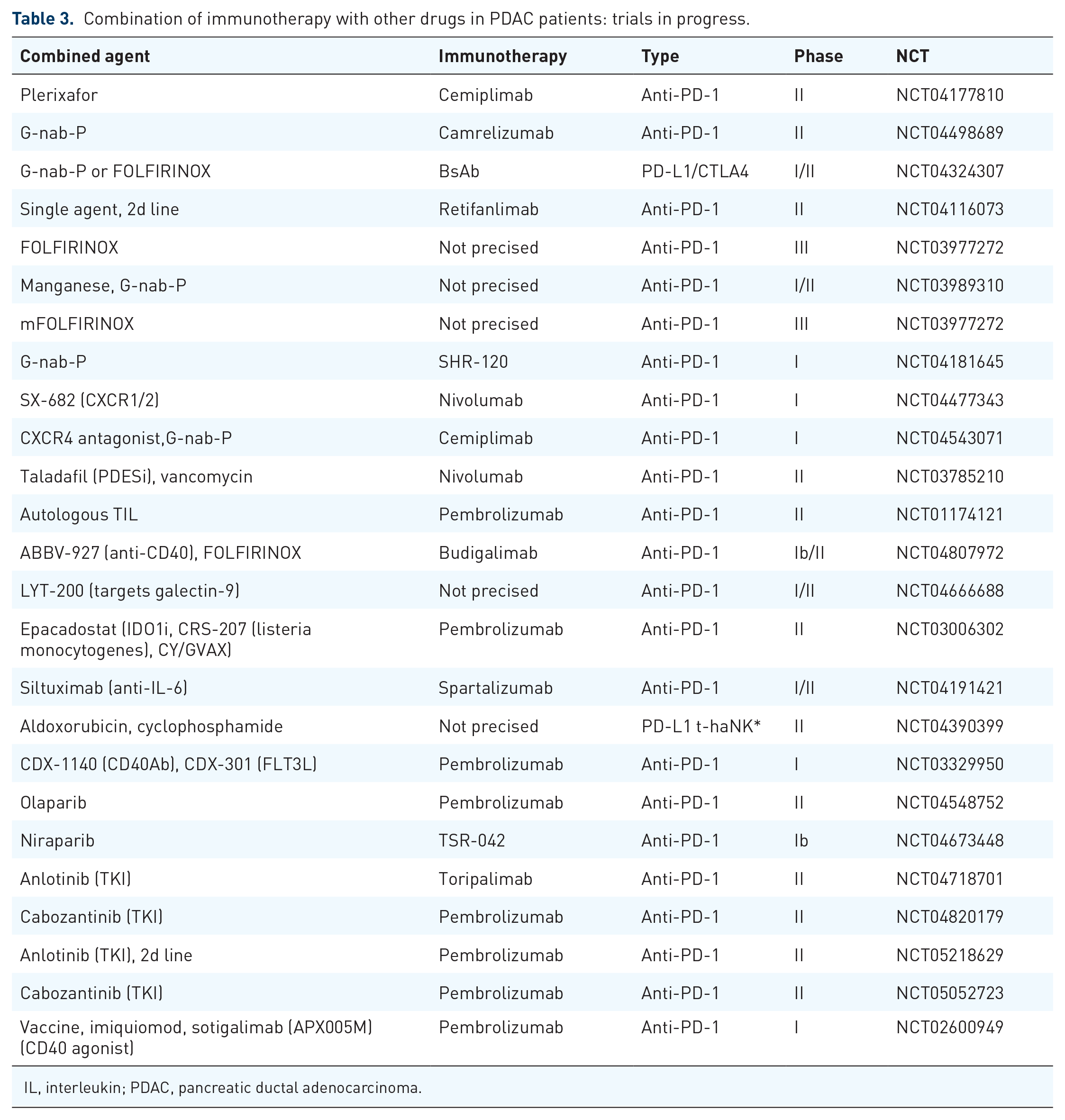
IL, interleukin; PDAC, pancreatic ductal adenocarcinoma.
Emerging targets of interest
Metabolic pathways
The harsh PDAC microenvironment explains why tumor cells must find alternative nutrient sources. 120 Tumor cells become dependent upon the activity of an oncogene for survival and proliferation via the mitochondrial metabolism and ATP production. This is mainly fueled by glutamine. 121 and KRAS signaling, which promotes extracellular glucose avidity and capture via upregulation of the glucose transporter GLUT1 and hexokinase. 21 This oncogene addiction may be the ‘Achilles’ heel’ of tumor cells and represent a potential therapeutic target. 22
Devimistat selectively inhibits the tricarboxylic acid cycle in the mitochondria of tumor cells by impairing pyruvate dehydrogenase and α-ketoglutarate dehydrogenase enzyme activity. Production of the anabolic intermediates required for effective DNA damage repair is decreased as a result of the impact on mitochondrial metabolism. In a phase I study, the combination of devimistat and modified FOLFIRINOX (mFOLFIRINOX) in 20 patients led to a high tumor response rate (61%). 122 However, the phase III study did not show any improved OS in the mFOLFIRINOX plus devimistat arm compared to FOLFIRINOX alone (HR = 0.95, median OS: 11.1 versus 11.7 months). 123
Constitutive KRAS signaling leads to addiction to metabolites such as glutamine and asparagine, used by non-canonical metabolic pathways.121,124 Glutamine deprivation and/or inhibition of enzymes downstream of KRAS result in suppression of PDAC cell growth. As PDAC are hypovascular tumors, enhanced asparagine synthetase expression is a key mechanism of adaptation to hypoxia and glucose deprivation.
125
Thus, modulation of glutamine and asparagine levels may be critical in these cells.
These findings add to the negative results from other trials targeting metabolism in PDAC with metformin, 129 evofosfamide, 130 or hydroxychloroquine (HCQ). 131 Nevertheless, other metabolic strategies are still being explored. Autophagy involves the degradation and recycling of damaged organelles and protein aggregates after autophagosome and lysosome fusion, which promote damaged cancer cell survival. 132 Autophagy is elevated in PDAC cells. 133 It promotes immune evasion by degrading the major histocompatibility complex class I (MHC-I) and decreasing tumor infiltration by CD8+ T cells. 23 MHC-I molecules are located in autophagosomes and lysosomes rather than on the cell surface and are targeted for degradation by an autophagy-dependent mechanism. Thus, inhibiting autophagy with drugs such as HCQ could sensitize PDAC cells to immune checkpoint blockade. 24 The ERK inhibitor LY3214996, which has shown some activity in BRAF- and KRAS-mutant models, administered as a single-agent or combined with HCQ (NCT04386057), and the paricalcitol, HCQ and gemcitabine-nab-paclitaxel combination (NCT04524702) have also been tested in two phase II trials in advanced PDAC patients.
The alpha-hydroxylated polyunsaturated fatty acid ABTL0812 induces sustained endoplasmic reticulum stress via induction of tribbles-3 pseudokinase expression, which results in inhibition of AKT-MTORC1 and dihydroceramide accumulation in human PDAC cell lines leading to autophagic activation and cell death.134,135 The combination of ABTL0812 with FOLFIRINOX is being tested for the first-line treatment of metastatic PDAC in a phase I-II trial (NCT04431258).
Stroma
Recombinant human hyaluronidase
PDACs have a dense and abundant stroma, which is known to be a critical mediator of disease progression through direct effects on cancer cells and indirect effects on the immune microenvironment of the tumor. The three major entities in the PDAC stroma are the extracellular matrix (ECM), vasculature (poorly developed in PDAC which are highly hypoxic), immune cells (mainly, pro-tumoral, inflammatory M2 and MDSC and low number of cytotoxic T cells), and cancer-associated fibroblasts (CAFs).136,137
PDAC is a highly chemoresistant cancer which may be because desmoplasia impedes drug delivery. This suggests that stromal modifying agents, such as pegvorhyaluronidase alfa (PEGPH20), an enzyme that temporarily degrades hyaluronic acid (HA), could decrease tumor pressure and vascular compression, thus penetrating the ‘halo’ surrounding the tumor cells. PEGPH20 has been developed and investigated in phase I-III studies. 138 This drug was assessed in a phase Ib/II clinical trial in combination with modified FOLFIRINOX (mFOLFIRINOX). A unexplained detrimental effect was reported in the PEGPH20 arm with a median OS that was nearly 50% lower (7.7 months versus 14.4 months in the chemotherapy alone group) and an increase in the rate of severe treatment-related adverse events (45% versus 9% in the mFOLFIRINOX alone arm). 139 Otherwise, in a phase III trial, the addition of PEGPH20 to gemcitabine plus nab-paclitaxel backbone chemotherapy did not improve survival. 140
Other ECM targets are currently under clinical investigation. 136 For example, the normalization of tumor vasculature by blocking the vascular endothelial growth factor (VEGF)–VEGF receptor 2 axis can improve pericyte coverage and tumor perfusion, leading to decreased hypoxia, increased drug delivery and CD8+ T-cell trafficking into the tumor through the action of the leukocyte adhesion molecules, intercellular adhesion molecule and vascular adhesion molecule. 141
Moreover, the PDAC TME has distinct CAFs populations. 142 There is an overwhelming body of evidence showing that CAFs are not mere cellular bystanders in cancer, but active players in the process of cancer initiation, progression, and metastasization. 143 CAFs have long been considered to be pro-tumoral cancer cell partners in PDAC.
These data have led to a paradigm suggesting that stroma and in particular CAFs promote PDAC tumor growth and metastases and contribute to the extensive resistance to therapies. This mechanistic model promoted the clinical development of anti-tumor strategies to deplete the stroma by disrupting the signaling pathways involved in intercellular communication between PDAC tumor cells and CAFs. These include the Sonic Hedgehog (SHH) or TGFβ pathways, or direct targeting of the ECM, such as HA – the main ECM component responsible for the elevated interstitial fluid pressure, or collagen I. Preclinical studies in murine models showed that SHH inhibition reduced stromal abundance, increased tumor perfusion, and thus, improved chemotherapy delivery. 144 However, phase II studies with SHH inhibitors in PDAC were negative. 145 Similarly, although several clinical trials, including the phase III MAESTRO study, have targeted TGFβ in a variety of cancers including PDAC, and which is overexpressed in 35% of PDAC, patient OS was not improved. 146 Melisi et al. 25 have presented preliminary results of a phase Ib study (NCT02734160) testing the TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab. DCR was of 25%.
Other ongoing trials with other agents such as bintrafusp (M7824) in a phase I/II trial plus stereotaxic body radiation therapy (NCT04327986) or LGK974 (NCT01351103) are being tested. As mentioned above, PEGPH20 has also been evaluated with negative results. 140 Preclinical studies further highlighted the deleterious effects of stromal depletion. Non-selective suppression of CAFs from the stroma, either by genetic αSMA-positive cell depletion 147 or by pharmacological inhibition of the SHH pathway, 26 adversely modulated immunity (increase in immunosuppressive regulatory T cells) and angiogenesis (increased tumor vascularization) in PDAC TME and led to undifferentiated, more invasive tumors.26,27 Moreover, deletion of the Col1a1 gene in αSMA-positive CAFs led to a significant decrease in total stromal collagen type I deposition and acceleration of disease due to the suppression of CD8+ T cells. 28 Overall, these negative results (i) highlighted the limited predictability of preclinical models and (ii) suggested that some CAF subpopulations or ECM components may play a protective role in blocking the progression of PDAC. Despite certain disappointing results in this poorly vascularized, hypoxic microenvironment, clinical trials targeting angiogenesis and CAFs subtypes are needed. 9
FAK
Nuclear focal adhesion kinase (FAK) regulates the transcription of chemokines that drive recruitment of tumor-associated regulatory T cells (Tregs), which contribute to an immunosuppressive TME in PDAC by inhibiting cytotoxic CD8+ T-cell activity. 148 Thus, FAK inhibition may modulate the cellular and molecular composition of immuno-suppressive TME. 29 On the other hand, stromal depletion by FAK inhibitor therapy may provoke treatment resistance through STAT3 signaling activation. 149 The results of a phase II trial combining GSK2256098 and trametinib (NCT02428270) in advanced PDAC patients were disappointing. 150 The FAK inhibitor defactinib is currently being tested in combination with the anti-PD-1, pembrolizumab and gemcitabine (NCT02546531, NCT02758857, and NCT03727880), or trametinib (NCT02428270).
BTK
Bruton tyrosine kinase (BTK) is a non-receptor enzyme from the Tec kinase family expressed in hematopoietic cells (B, myeloid and mast cells, and platelets). BTK-dependent signaling in mast cells and myeloid cells plays an important role in peri-tumoral inflammatory stroma. Ibrutinib specifically inhibits the release of IL-8, MPC-1, and tumor necrosis factor α from mast cells leading to decreased fibrosis and extended survival, and improves the response to clinical standard-of-care therapy in mouse models. 151 In the phase III RESOLVE trial, the addition of ibrutinib to nab-paclitaxel plus gemcitabine failed to improve OS (9.7 versus 10.8 months; p = 0.3225) and provided an even shorter PFS (5.3 versus 6.0 months; p < 0.0001) in the ibrutinib arm than in the placebo arm. 152 The efficacy of the combination of ibrutinib with the anti-PDL-1 durvalumab, 153 and acalabrutinib alone or combined with pembrolizumab 154 was found to be limited in PDAC patients.
CTGF
Connective tissue growth factor is strongly expressed in CAFs and tumor cells. Pamrevlumab (FG-3019) enhances the chemotherapy response without increasing drug delivery in murine ductal pancreas cancer by decreasing expression of the X-linked mammalian inhibitor of apoptosis protein gene. 155 In a phase I/II trial, patients with locally advanced PDAC who received a combination of gemcitabine/nab-paclitaxel plus pamrevlumab achieved surgical resection in 33% of cases versus 8% in those with gemcitabine-nab-paclitaxel alone. 156 The combination of pamrevlumab plus gemcitabine-nab-paclitaxel or FOLFIRINOX has been evaluated in patients with advanced PDAC in a phase III trial and the results are pending (NCT03941093 and NCT04229004).
Tumoral and stromal signatures
The choice of systemic therapy using combined, potentially toxic drugs is mainly based on the patient’s performance status and biological/liver tests. Thus, diagnostic tests should be used to guide the choice of individualized therapy for patients. 157 Combined genomic, transcriptomic, and therapeutic profiling using patient-derived organoids (pharmacotyping) could help identify molecular and functional subtypes of PDAC for this purpose. 158 ORGANOTREAT-01 (NCT05267912) is a clinical trial establishing organoid models from the patient’s biopsy to test the chemosensitivity of available drugs (‘chemogram’). Even if pharmacotyping is promising as surrogate tumor models for personalized oncology, this strategy has several limitations for a highly aggressive disease such as PDAC: (1) because the delay required for pharmacotyping is long (usually several weeks), patients may receive treatments that modify tumor sensitivity; 2) in non-experienced centers the quality of the organoid cultures may be suboptimal (<60%) because this approach is technically challenging. On the other hand, the predictive multigene signatures based on RNA expression measurement can be obtained rapidly from patient biopsies in breast, prostate, and pancreatic cancers.159–163 In practice prediction of drug sensitivity despite the lack of defined molecular targets seems to be more useful than predicting the risk of recurrence. The predictive value of transcriptomic signatures in PDAC, in particular the basal-like and classic epithelial subtypes, is of great interest. 164 In the COMPASS trial (NCT02750657), Aung et al. 165 identified 30% of actionable mutations (ARID1A, BRAF, CDK4/6, PIK3CA PTEN, and RNF) in their series of patients with PDAC. The efficacy of modified (m)FOLFIRINOX and gemcitabine-nab-paclitaxel combinations was assessed according to the molecular subtype of the tumor on RNA-sequencing.. ‘Basal-like’ and ‘classic’ modified Moffitt subtypes were identified in 20% and 80% of cases, respectively. The basal-like subtype was associated with a lower objective response rate than the classic phenotype in patients who received mFOLFIRINOX (10% versus 33%, respectively, p = 0.02) and median OS (5.9 versus 9.3 months, respectively, HR = 0.47; 95% CI: 0.32–0.69; p = 0.0001). 166 Nevertheless, no alternative to mFOLFIRINOX has been found to be more effective for the basal-like subtype. Another transcriptomic signature is the Pancreatic Adenocarcinoma Molecular Gradient. Unlike all other stratification schemas, this signature classifies PDAC along a continuous gradient with a prognostic value that also predicts the response to mFOLFIRINOX.162,163 GATA6 RNA expression is lower in basal-like tumors. GATA6 acts as a tumor suppressor by blocking KRAS G12V-driven pancreatic tumorigenesis and enforcing differentiation. GATA6-low cell lines derived from patient-derived xenografts were resistant to 5-fluorouracil but not gemcitabine patient-derived organoids.158,167
Nicolle et al.162,163 have identified a RNA-based whole transcriptome signature that predicts sensitivity to gemcitabine (‘GemPred’) in the adjuvant setting after surgical resection of PDAC. 168 Among gemcitabine-treated patients, OS was significantly higher in GemPred-positive patients than in GemPred-negative patients [91.3 months (95% CI: 61.2–not reached) versus 33 months (95% CI: 24–35.2); HR 0.403 (95% CI: 0.221–0.735, p = 0.00216), respectively]. Genome-wide RNA profiles were also obtained from routine formalin-fixed, paraffin-embedded samples of fine-needle aspirate material. This is valuable for routine use, as pancreatic biopsies are performed using fine-needle aspiration under endoscopic ultrasound control. 168 Finally, this signature could help reposition gemcitabine in frail PDAC patients who are not eligible for FOLFIRINOX, whatever the indication.
High replicative stress
Genomic instability is a key hallmark of cancer, occurring due to defects in the DNA damage response and/or increased replication stress.169,170 ATR and ATM are key components to the DNA damage response and are prime targets for DNA damage response inhibitors, because of their central regulatory function in activating the response to both single-strand breaks and DSBs. Both of these proteins work through distinct but overlapping pathways to halt the cell cycle and initiate DNA damage response pathways. 169
There is an interest in using ataxia–telangiectasia and ATR protein kinase inhibitors in cancer, especially in high replicative stress cancers. ATR inhibition leads to replication fork collapse as well as the loss of the G2-M checkpoint in cancers with increased levels of replication stress (defined by slowing or stalling of replication forks, e.g. with TP53 loss or CCNE1 amplification), allowing cells with damaged DNA to progress prematurely into the M phase, leading to mitotic catastrophe and cell death.30,171–173 Thus, ATR inhibition causes unresolved replication stress that leads to the induction of DNA DSBs causing cell death. 174 ATR inhibitors such as berzosertib have already been evaluated in preclinical studies. 175 Most PDAC display high replicative stress. A recent study based on whole exome sequencing datasets reported that the ‘squamous’ transcriptomic subtype of PDACs shows signs of high replicative stress, which may make them sensitive to ATR inhibitors. The authors of this study showed that a novel DNA replication stress transcriptomic signature predicts the response to new ATR and WEE1 inhibitors. High replication stress and the presence of DNA repair gene deficiencies appear to be distinct entities, which may exist independently and could be targeted by different drugs. 176
In particular, this ‘squamous’ subtype is enriched in TP53 and KRAS mutations. Indeed, tumor cells with TP53 mutations (a major cell cycle checkpoint) are subject to DNA damage, while those with KRAS mutations are exposed to replicative stress.171,177 Thus, replicative stress may be increased in the presence of a double KRAS and TP53 mutation, which is found in more than 50% of pancreatic cancers.
Others
CAR-T-cell development
Adoptive T-cell therapy, which redirects a patient’s own T cells against a tumor antigen using CAR gene transfer technology, has been remarkably successful in hematological tumors in the past several years. 178
The complex TME of PDAC, as well as the stromal obstacle limiting the immune response, and the expression of checkpoint blockade on T cells are obstacles to this approach. 31 CAR-T cells face major difficulties due to the aggressiveness of the disease and the usually late stage diagnosis with extensive tumor dissemination. CAF-depletion, remodeling approaches using CAR-T cells or pharmacological substances such as ATRA or nab-paclitaxel might help limit the influence on the TME. 31
Some of these obstacles may be overcome or turned into a specific targeting strategy, and clinical trials to overcome the physical and environmental barriers in the TME are ongoing. 179
Target antigens in these trials include mesothelin, prostate stem cell antigen, carcinoembryonic antigen (CEA), HER2, MUC1, and CD133. The best results from clinical trials in CAR-T cells in PDAC have been obtained from mesothelin 180 and CEA targets. 181
Mesothelin is expressed in 80% of PDACs,182,183 and 25%–100% of the cells in positive tumors express the antigen. 184 Positive results in a phase I study in chemotherapy-refractory metastatic PDAC provide evidence for the potential antitumor activity of autologous mesothelin-specific CAR-T cells, as well as PDAC resistance to the immune response. 180 A more recent phase I trial investigated the safety and activity of lentiviral-transduced CAR-T-meso in 15 patients with malignant pleural mesothelioma, ovarian carcinoma, and PDAC. Patients with PDAC showed the greatest percentage of lesions with a disease progression within 28 days. 185 Other results in patient-derived xenograft models from patients with stage IV PDAC suggest that a switchable CAR-T system to target the antigen HER2, upregulated on tumor cells is effective against aggressive, disseminated PDAC. 186 This strategy is ambitious because historically, when a CAR-T HER2/NEU was tested, the patient died from cardiopulmonary toxicity within days. 187 The CEA is a glycoprotein expressed in nearly 75% of pancreatic cancers 188 and it can also be targeted in PDAC. There are ongoing trials evaluating the role of CAR-T cells activated with the CEA for this indication. 189 While there are no reports of pancreatic cancer-specific CAR T-cell trials targeting CEA, there are preliminary results from a phase I study evaluating the feasibility of delivering first-generation CAR-T-cell therapy to patients with advanced CEACAM5+ tumors. No objective clinical responses were observed and this trial was stopped due to pulmonary toxicity. 190 A recent trial identified CEACAM7 (CGM2), a member of the CEA family of proteins expressed in a large subset of PDAC tumors, as a potential CAR T-cell target for PDAC. CAR-T cells targeting CEACAM7 can target antigen-expressing tumor cells, and mediate remission in patient-derived xenograft tumors. 181 Thus, clinical trials are ongoing. Recently, Leidner et al. reported the results of a patient who was treated with her own autologous T cells that were genetically engineered to clonally express two allogeneic HLA-C*08:02–restricted T-cell receptors (TCRs) targeting the mutant KRAS G12D expressed in her tumor. This resulted in an impressive regression of PDAC metastases. These very promising results suggest that TCR gene therapy targeting the KRAS G12D hotspot mutation merits further study in prospective clinical trials. 102
Restoring the immunity by vaccines
Although cancer vaccines have been evaluated in clinical settings and an antitumor immune response has been shown in PDAC, 191 there is no clear proof that they will be used in clinical practice in the near future. 192
GVAX, an irradiated allogeneic whole tumor cell vaccine in which PDAC cells are engineered to express granulocyte-macrophage colony stimulating factor (GM-CSF), was evaluated in a small study including 30 previously treated PDAC patients. 193 Disease control with the combination of GVAX and CPIs was promising in that study. 193 In another more recent study, GVAX and ipilimumab maintenance therapy did not improve OS compared to the continuation of chemotherapy and resulted in poorer survival in metastatic PDAC. However, clinical responses and biological effects on immune cells were observed. 194 Therapeutic cancer vaccines also include DC, DNA, and peptide vaccines that provoke the presentation of immunogenic cancer antigens to the immune system, resulting in activation of cancer antigen-specific CTLs in vivo (and a subsequent anti-cancer immune response). 191 In this context, the results of mesothelin, Mucin-1 (MUC1) and telomerase-based vaccine trials were disappointing. 192 For example, tolerance was good and some efficacy was shown in a phase I/II trial of a combination of telomerase peptide GV1001 with GM-CSF for nonresectable PDAC. 195 However, after these promising results, no benefit was found in a phase III trial evaluating a combination of chemotherapy with a telomerase vaccination (GV1001 or TeloVac). 196 In another recent study, a new concept was proposed involving screening of PDAC antigens to develop an mRNA vaccine. A series of targetable antigens including ADAM9, EFNB2, MET, TPX2, TMOD3, and WNT7A were found to be promising mRNA vaccine candidates. 197 Therapeutic cancer vaccines targeting KRAS were also developed to elicit a KRAS-specific immunological response and seem to be promising and safe with some clinical benefit. 198 Although results are not highly encouraging several vaccine trials are still ongoing to evaluate non dMMR/MSI-H PDAC immune responders, either whole cell or peptide-vaccines, according to HLA-A2 status (TEDOPAM-PRODIGE63 trial, NCT03806309).
Role of microbiota
Oral and gut microbiota, the intratumoral microbiome, that is, the bacterial and fungal microorganisms present in the tumor, has recently been shown to be a new partner of PDAC TME, modulating pancreatic carcinogenesis, intratumoral immune infiltrates, and the response to chemotherapy. 199 Geller et al. 200 showed that intratumoral gammaproteobacteria (e.g. Klebsiella pneumoniae, Escherichia coli K12, Mycoplasma Hyorhinis) expressing long isoforms of CDA, an enzyme involved in the metabolism of gemcitabine, were found in 76% of resected PDAC samples. These bacteria could degrade gemcitabine in vitro, and induced therapeutic resistance to this drug in a mouse colorectal cancer model. Resistance to gemcitabine was suppressed when this enzyme was deleted or by co-treatment with ciprofloxacin. Thus, bacteria might be responsible for the poor clinical response to gemcitabine in PDAC. 200 In addition, Nejman et al. 201 found that intratumoral bacteria are predominantly intracellular and present in both cancerous and immune cells (macrophages and others). In tumor tissue, the correlation between bacterial species or their predicted functions and response to immune therapy have been established in a large spectrum of tumors. A retrospective study of 211 patients with PDAC supported this hypothesis showing that adjuvant treatment with gemcitabine improved PFS in patients without Klebsiella pneumoniae in the bile culture. Similarly, quinolone administration improved OS in patients with quinolone-sensitive Klebsiella pneumoniae. 202
Based on this evidence, manipulation of the intratumoral microbiota is emerging as a potential new therapeutic approach to PDAC. To enhance their efficacy, these strategies are usually developed in combination with other therapeutic options such as chemotherapy or immunotherapy. Among them, fecal microbiota transplantation, diet-based interventions, antibiotics, and probiotics are under investigation. 203
Two studies are currently recruiting to explore the role of gut microbiota (PDAC patients or healthy controls) on anti-MSLN (mesothelin protein) CAR-T treatment (NCT04203459) or fecal microbiota transplantation (in patients with resectable PDAC) (NCT04975217).
Conclusion
The number of actionable molecular targets for PDAC is significantly increasing, offering promising potential treatments and emphasizing the importance of continued intensive clinical and basic research in this field. Olaparib paved the way in the subset of patients with BRCA 1/2 germline mutations. Somatic analysis of tumors by next generation sequencing is now more accessible allowing a theragnostic approach to this rapidly progressing disease. The effective targeting of KRAS mutations is clearly a major challenge because this gene drives up to 90% of PDAC and explains most cases of therapeutic resistance. Although targeting metabolic pathways is theoretically promising, results have been disappointing until now. The stroma signatures may help improve selection of patients for a given chemotherapy in the near future. While immunotherapies are not effective as single agent, new combinations with other drugs or T-cell engineering may bring them to the forefront once again. Finally, these promising trends in the therapeutic landscape are indissociable from all aspects supportive care.