
Abstract
Maintenance of genomic stability is a critical determinant of cell survival and relies on the coordinated action of the DNA damage response (DDR), which orchestrates a network of cellular processes, including DNA replication, DNA repair and cell-cycle progression. In cancer, the critical balance between the loss of genomic stability in malignant cells and the DDR provides exciting therapeutic opportunities. Drugs targeting DDR pathways taking advantage of clinical synthetic lethality have already shown therapeutic benefit – for example, the PARP inhibitor olaparib has shown benefit in BRCA-mutant ovarian and breast cancer. Olaparib has also shown benefit in metastatic prostate cancer in DDR-defective patients, expanding the potential biomarker of response beyond BRCA. Other agents and combinations aiming to block the DDR while pushing damaged DNA through the cell cycle, including PARP, ATR, ATM, CHK and DNA-PK inhibitors, are in development. Emerging work is also uncovering how the DDR interacts intimately with the host immune response, including by activating the innate immune response, further suggesting that clinical applications together with immunotherapy may be beneficial. Here, we review recent considerations related to the DDR from a clinical standpoint, providing a framework to address future directions and clinical opportunities.
The DNA damage response
Genome instability is described as one of the hallmarks of cancer. 1 Human DNA is continuously exposed to potential sources of damage, both exogenous – such as ultraviolet and ionizing radiations, chemicals and chemotherapeutics – and endogenous – such as reactive oxygen species and faulty DNA replication.2,3 Cells have evolved a complex DNA damage response (DDR), which is in charge of repairing DNA damage and promoting the maintenance of genome integrity. Defects in DDR are associated with increased mutational load and genome instability and are a well-recognized cause of neoplastic transformation and proliferation. Cells harbouring DDR defects can become reliant on other repair pathways for survival, which makes DDR targeting an attractive therapeutic strategy.1,2
Mechanisms of DDR are numerous and partially overlap. Their functioning involves multiple sensors of damage, signalling factors that activate cell-cycle checkpoints and effector proteins of repair. This orchestra is responsible for processing the two main types of DNA lesions: single-strand breaks (SSBs) and double-strand breaks (DSBs) (Figure 1). 4 If DNA is not repaired, replication stress results. Replication stress is slowing of the DNA replication fork. 5 It is a highly dynamic chain of events starting from acutely arrested forks with fully assembled replisomes, uncoupling of the DNA helicase and polymerase, RPA coated ssDNA and ATR activation. 5 If replication stress persists, stalled forks are converted into ‘collapsed forks’ with dissociation and/or impaired modifications of replisome components.6,7 Further extension of replication stress converts forks to DNA DSBs, causing replication catastrophe and cell death. 8

DDR pathways. (a) DNA strand breaks activating DNA repair pathways; (b) stalled replication fork leading to ATR activation.
SSBs can result from endogenous oxidative damage, defective activity of cellular enzymes or erroneous incorporation of ribonucleotides in DNA. 9 Repair can occur through base excision repair (BER), nucleotide excision repair (NER) and mismatch repair (MMR). 10 BER is involved in the removal of small base adducts. Poly (ADP-ribose) polymerase-1 and 2 (PARP1 and PARP2) are crucial proteins for BER, acting as sensors of SSB and promoting the recruitment and activation of critical downstream SSB repair effectors such as XRCC1. 9 NER is engaged in the repair of lesions that cause a distortion of the DNA helix, generally as a result of UV-induced damage. It promotes the removal of short oligonucleotides, involving ERCC1 as one of the key effector proteins.11,12 The MMR system is responsible for correcting base–base mispairing and insertion/deletion loops that can occur during DNA replication. These faulty areas of DNA are recognized by the proteins MSH2, MSH3 and MSH6, which recruit MLH1 and PSM2 on the sites of damage, enabling repair. 13 Unrepaired SSBs can lead to cell death or to the collapse of DNA replication forks, with the formation of DSBs. 9
DSBs are among the most destructive DNA lesions. Repair occurs through homologous recombination (HR), non-homologous end joining (NHEJ) and single-strand annealing (SSA). HR is an accurate process taking place during the S and G2 phases of the cell cycle, and promoting precise repair of the damaged area of DNA by using the sister chromatid as a template. Several genes with a tumour-suppressor activity are crucial to this process, including BRCA1, BRCA2, PALB2 and RAD51, and their functioning is essential for an error-free repair. 14 Defective HR cells are redirected towards more error-prone DSB repair pathways such as NHEJ, which occurs throughout the whole cell cycle, and SSA. In this circumstance, repair is performed by direct ligation of the two DNA broken strands, which can lead to DNA loss and increased mutagenic potential. 15
Defects in the components of the DDR network drive a variety of hereditary and sporadic tumours. Loss-of-function mutations in the MMR system are associated with the formation of repeated DNA sequences, a phenotype known as microsatellite instability (MSI). MSI can result from mutations of MMR genes at the germline level which are characteristic of the hereditary non-polyposis colorectal cancer (HNPCC or Lynch syndrome) or from somatic mutations or epigenetic silencing through methylation of MLH1 promoter. Somatic loss of MMR components has been described in 15% of sporadic colorectal cancers as well as in other non-colorectal cancers, particularly endometrial cancer. 16 Aberrations in HR genes correlates with genome instability and cancer susceptibility. Loss of function in the genes BRCA1 and BRCA2 is a predisposing factor for the development of hereditary and sporadic cancers: ovarian, breast, pancreatic and prostate cancers.17,18 BRCA1/2 deficiency can arise from germline or somatic gene mutations or from BRCA1/2 epigenetic silencing. Methylation of the BRCA1 promoter is described in 11–14% of sporadic breast cancers and 5–31% of ovarian cancers.19,20 A subset of sporadic tumours has been found to share common features with BRCA-deficient tumours by means of mutation or epigenetic deregulation of genes involved in the HR, including RAD51, RAD54, DSS1, RPA1, NSB1, ATR, ATM, CHEK1, CHEK2, FANCD2, FANCA and FANCC 2.21,22 Data from large-scale genome analysis in ovarian cancer from The Cancer Genome Atlas (TCGA) report an incidence of 51% of alterations in HR genes in 316 samples analysed. 23 Sequencing of metastatic castration-resistant prostate cancers has revealed aberrations in key DDR genes in up to 23% of cases. 24
With the widening use of next-generation sequencing techniques, the plethora of DDR defects being detected is ever increasing, focusing efforts in developing effective therapeutic strategies to target these.
Targeting DDR defects through synthetic lethality
DNA damage has for years represented a backbone of cancer treatment. Platinum compounds such as cisplatin and carboplatin, by inducing interstrand or intrastrand cross-linking, cause double helix disruption leading to DNA damage. Despite their activity in a broad spectrum of cancers, treatment resistance develops through a number of mechanisms which can involve upregulation of DDR components. 25 DDR inhibitors have been developed, and as a first approach were tested in combination with platinum agents. However, the overlapping toxicity profile represented a challenge. 26 Testing as single agents has followed based on the observation of synthetic lethality occurring between two or more DDR components in specific molecular contexts. 27 Two genes are considered synthetic lethal when cell viability is conserved if either of them is inactive, whereas the impairment of both results in cell death. 28 Cancer cells with defects in one DDR pathway often depend on other pathways for their survival, and targeting these pathways of reliance can be exploited to cause selective cancer cell death.
PARP inhibitors
The paradigm in this field is represented by the development of PARP inhibitors in BRCA1/2-defective cells. 4 PARP1 and PARP2 are key sensors of DNA damage and are crucial in activating the cascade of SSB repair and BER. Their inhibition causes an increase in DSBs, which are normally repaired by HR. In cells harbouring defects in the HR system, such as BRCA1/2 mutant cells, inhibition of PARP enzymes results in cell-cycle arrest and apoptosis of cancer cells through synthetic lethality.29,30
The clinical application of PARP inhibitors is most advanced in ovarian cancer, where the PARP inhibitor olaparib has received regulatory approval in a number of settings. In a phase II study of 57 patients with BRCA1/2-mutant ovarian cancer treated with olaparib 400 mg twice per day, Audeh and colleagues have reported an overall response rate of 31%. 31 A phase II trial enrolled 64 patients with high-grade serous ovarian cancers and demonstrated an overall response rate of 41% in the BRCA1/2-mutant group. 32 Based on these findings, olaparib received accelerated approval from the FDA in 2014 for fourth-line or later treatment of germline BRCA1/2-mutant ovarian cancer. Subsequent trials have investigated the role of olaparib as maintenance therapy after platinum-based chemotherapy with subsequent FDA approval in the platinum-sensitive, non-BRCA-mutated setting. In a randomized placebo controlled phase II trial by Ledermann and colleagues, olaparib significantly improved progression-free survival (PFS) in 265 patients with recurrent platinum-sensitive high-grade serous ovarian cancer [median PFS 8.4 months versus 4.8 months; hazard ratio 0.35; p < 0.001]. A subgroup analysis of the study has reported that the benefit of maintenance olaparib was increased in the BRCA1/2 mutant sub-population (median PFS 11.2 months versus 4.3 months; hazard ratio 0.18; p < 0.000). 33 This trial has led to the European Medicines Agency (EMA) approval of olaparib in BRCA1/2-mutant or platinum-sensitive ovarian cancer (regardless of BRCA status) as maintenance after complete or partial response to platinum-based chemotherapy. The SOLO II phase III trial of 295 patients with platinum-sensitive BRCA1/2-mutant ovarian cancer, pretreated with at least two lines of chemotherapy, saw a significant PFS benefit of olaparib compared to placebo (19.1 months versus 5.5 months; hazard ratio 0.30; p < 0.0001), which has led to approval by the FDA for the tablet formulation in this setting. 34
Olaparib has shown encouraging activity in a phase II trial in 27 patients with BRCA1/2-mutant advanced breast cancer, which has reported an overall response rate of 41%. 35 More recently, results from the randomized phase III trial OLYMPIAD compared olaparib versus standard chemotherapy in patients with germline BRCA-mutant HER2-negative breast cancer treated with two or fewer chemotherapy lines. Among the 302 patients enrolled, olaparib showed a significant benefit in PFS (7.0 months versus 4.2 months; HR for disease progression or death, 0.58; p < 0.001). 36
Olaparib has also proven remarkable activity in DDR-defective metastatic castrate-resistant prostate cancer (mCRPC), which represent up to 23% of all prostate cancer cases. 24 Mateo and colleagues conducted a phase II trial (TOPARP-A) of olaparib 400 mg twice daily in unselected mCRPC patients pretreated with docetaxel and abiraterone and/or enzalutamide. Among the 49 patients enrolled, a response by the composite endpoint (comprising RECIST 1.1, PSA or CTC count) was reported in 16 (33%) patients, including PSA decline greater than 50% in 11 patients and 6 radiologic partial responses. Notably, the investigators performed next-generation sequencing on all patients enrolled, which has identified homozygous deletions or deleterious mutations in DNA repair-related genes in 16 out of 49 (33%), including BRCA1/2, ATM, PALB2, FANCA and CHK2. Among these DDR mutation carriers olaparib showed significantly increased activity, with responses occurring in 14 out of 16 (88%) patients. This trial has granted olaparib breakthrough therapy designation approval in BRCA1/2- or ATM-mutant mCRPC patients pretreated with one line of taxane chemotherapy and a new-generation antihormonal agent. The study further corroborates the strong rationale in developing PARP inhibition in DDR-defective patients beyond BRCA mutations. The second stage of the trial (TOPARP-B) is currently ongoing and prospectively recruiting patients carrying a DDR-defective signature to validate PARP inhibition activity in this subgroup. 37
Other PARP inhibitors have now reached the late stages of clinical development: rucaparib (AG014699; Clovis), talazoparib (BMN637; Medivation), veliparib (ABT-888; AbbVie) and niraparib (MK4827; Tesaro). Of note, rucaparib has received breakthrough therapy designation in BRCA1/2-mutant ovarian cancer progressing on two lines of platinum regimens based on the PFS benefit reported in the phase III trial ARIEL-2; 38 niraparib (MK4827; Tesaro) has been approved by the EMA and FDA as maintenance treatment of recurrent platinum-sensitive ovarian cancer, having showed activity in both BRCA-mutant and wildtype patients in the NOVA trial. 39
Single-agent activity of PARP inhibitors in non-HR-defective tumours has so far been limited; combination strategies of PARP with DNA-damaging cytotoxic agents could enhance sensitivity to PARP inhibition and a number of trials addressing this question are currently under way (Table 1). 40 Furthermore, acquired resistance to PARP inhibition is common. As observed with platinum agents, modifications in DDR pathways can occur through activation of NER, MMR and/or HR pathways, allowing for increased repair and promoting treatment resistance. 41 Similarly, increasing evidence is revealing a number of mechanisms underlying resistance to PARP inhibition. 42 Several mechanism of resistance have been proposed, among which the restoration of BRCA function through secondary frameshift mutations is the most well established. 43 Restoration of HR function by somatic mutations confers olaparib resistance.44,45 Combination of PARP inhibitors with other DDR agents, potentially exploiting DDR synthetic lethalities, or with chemotherapeutic agents are currently explored approaches in trying to overcome PARP inhibitor resistance.42,46 In the era of new DDR agents, treatment resistance will have to be taken into account.
PARP inhibitor phase II and III trials.

ATM inhibitors
ATM is a key protein in HR repair of DSB via HR. ATM acts as a signalling protein with hundreds of downstream substrates, including CHK2, a cell-cycle checkpoint activator. In preclinical studies, ATM inhibitors have sensitized cells to ionizing radiation and DSB-inducing agents; early-phase clinical testing of ATM inhibitors is currently ongoing. 47 ATM has a synthetic lethal relationship with PARP1 and preclinical models exhibit enhanced sensitivity to PARP inhibition of ATM-deficient cells. 48 Synthetic lethality exists also between ATM and ATR and between both ATM and ATR with XRCC, a relevant component of SSB repair through BER. 49
ATR inhibitors
ATR is an essential DDR kinase activated in response to replication stress and stalled replication forks. Through activation of multiple downstream effectors of which CHK1 and Wee1 are the most well characterized, ATR signalling promotes cell-cycle control and DNA repair through HR. Cancer cells, which harbour high levels of replication stress, are more likely to rely on the ATR pathway for survival. 50 Among agents in clinical testing, VX-970 (Vertex Pharmaceutical; now M6620, Merck), an intravenous ATR inhibitor, has shown target modulation and meaningful tumour control in a phase I trial as a single agent and in combination with carboplatin, including tumour responses in a patient with an ATM-deficient colorectal cancer and a patient with a BRCA1-mutant, platinum- and PARP-inhibitor-resistant high-grade serous ovarian cancer.51,52 Early-phase combination trials of VX-970 with gemcitabine and cisplatin exhibited encouraging activity results.53,54 Another oral ATR inhibitor, AZD6738 (Astrazeneca), has shown promising preclinical activity and is currently being tested in phase I trials as monotherapy or in combination with olaparib, radiotherapy regimens, carboplatin and immunotherapy agents (Table 2).4,55 ATR inhibition has been found to be synthetic lethal, with a number of DDR components in preclinical models including ERCC1, 56 XCRCC1, 49 CHK157 and ATM, 27 which could serve as a background for new clinical trials.
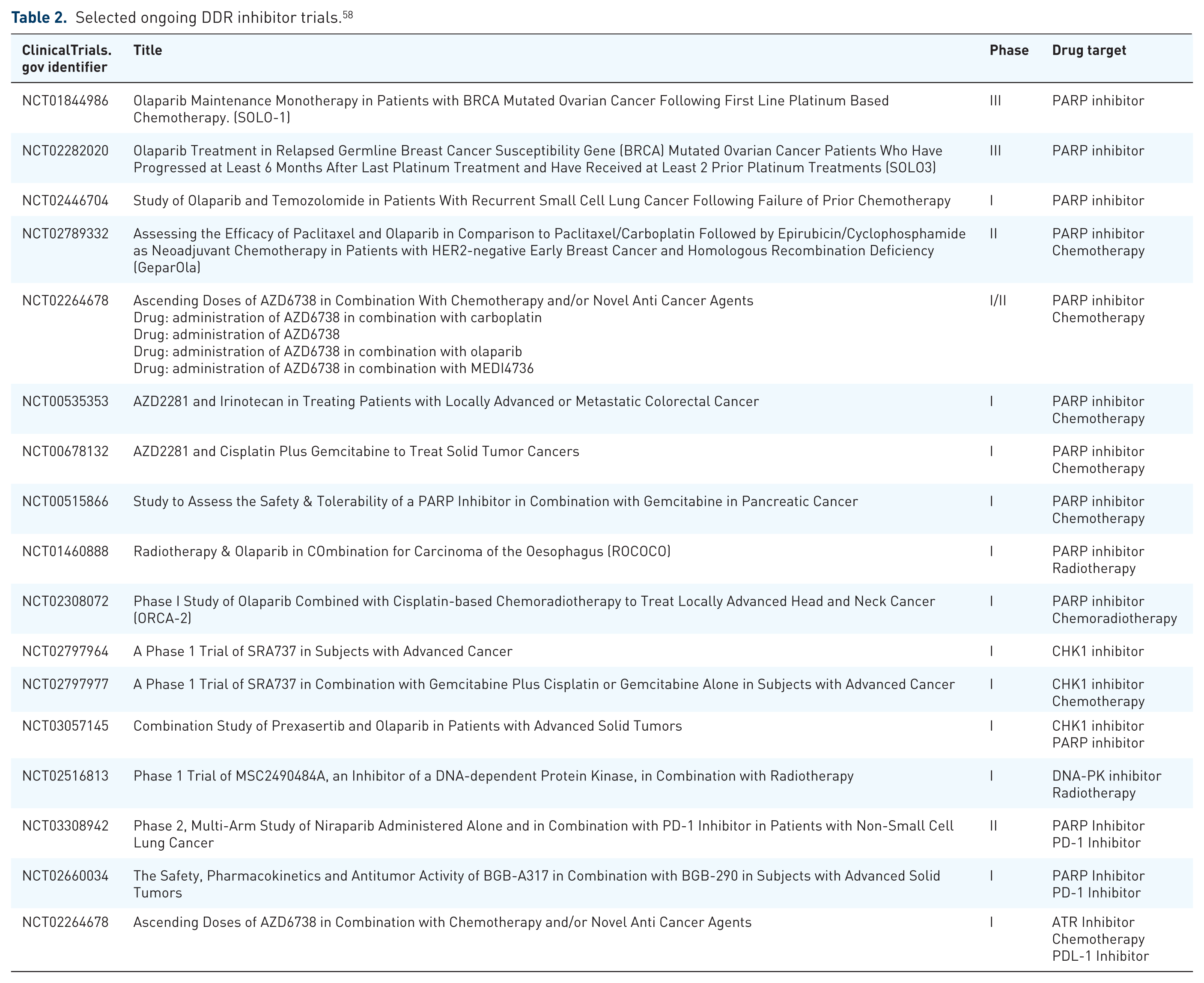
Selected ongoing DDR inhibitor trials. 58
DNA-PK inhibitors
DNA-PK (DNA-dependent serine/threonine protein kinase catalytic subunit) acts as a sensor of DNA damage and is crucial for repair through NHEJ. High levels of DNA-PK are correlated to poor prognosis and resistance to chemotherapy and radiotherapy in several tumour models.59,60 The inhibition of DNA-PK through small molecules has proven particularly active when combined with agents inducing DSBs, such as radiotherapy and topoisomerase inhibitors. Clinical studies involving DNA-PK inhibitors as a single agent or in combination with chemotherapy or radiotherapy are currently ongoing in patients with solid and haematological neoplasms (Table 2).61,62
CHK1 inhibitors
CHK1 is the most important phosphorylation target of ATR and mediates DNA repair and checkpoint activation. 50 CHK1 inhibitors are currently undergoing clinical testing both in combination with cytotoxics and as single agents. Among them, LY2606368 (Eli Lilly) as monotherapy has shown promising signs of anti-tumour activity and safe toxicity profile in a phase I trial, 63 and combination trials with cytotoxics are ongoing. Other CHK1 inhibitors currently in early-phase trials are GDC-0575 and SRA737, which are being tested as single agents and in combination with enhancers of replication stress (Table 2). Preclinical studies have reported a synergistic effect of combination of CHK1 inhibitors with PARP inhibitors and a trial investigating this strategy is now recruiting (Table 2).64,65
ATR and CHK1 activity is closely interrelated and there is an established synthetic lethal relationship between the two, which provides a rational for combination therapies of ATR and CHK1 inhibitors. 57 CHK1 inhibition has been found to be synthetic lethal with the Wee1 kinase in preclinical models (see below).66,67
Wee1 inhibitors
Wee1 is a key controller of cell-cycle checkpoint, particularly at the G2 phase; when activated by DNA damage it prevents progression of the cell to mitosis, allowing time for repair. 68 The Wee1 inhibitor AZD1775 (MK1775; Astrazeneca) exhibited single-agent activity as well as in combination with cytotoxic agents (gemcitabine, carboplatin, cisplatin) in early-phase trials and a favourable safety profile. Interestingly, two partial responses were observed in BRCA1-mutant patients, suggesting a role for Wee1 inhibition in HR-defective cancers.69,70 AZD1775 has undergone testing in a phase II trial in combination with carboplatin in patients with p53 mutant platinum-resistant ovarian cancers and has shown an overall response rate of 43%. 71 A phase II trial of AZD1775 in 121 ovarian cancer patients with platinum-sensitive disease randomized patients to AZD1775 in combination with paclitaxel-carboplatin or paclitaxel-carboplatin alone. A small PFS benefit was seen with a PFS of 34.14 versus 31.85 weeks (hazard ratio 0.63; p = 0.080). 72
Biomarkers of DDR inhibitors
As demonstrated by the clinical development of PARP inhibitors to date, the clinical utility of DDR inhibitors relies on establishing biomarkers of response to allow appropriate patient selection. Many putative biomarkers reflect aberrations in DDR pathways or genomic signatures that result from DNA damage. 73
Clinical biomarkers
Sensitivity to platinum-based chemotherapy is often taken as a surrogate biomarker of ‘BRCA-ness’ and thus sensitivity to PARP inhibitors. The FDA has approved platinum sensitivity as a biomarker for olaparib in the maintenance setting. However, this clinical biomarker is limited; some patients who respond to platinum do not respond to PARP inhibitors (for example, due to NER mutations) and some whom are resistant to platinum respond to PARP inhibition (for example, due to loss of TP53BP1 or REV7).32,74–76
Genomic biomarkers
As discussed, olaparib’s EMA (though not FDA) approval for treatment of ovarian cancer mandates germline BRCA1/2-mutant ovarian cancer and BRCA1/2 or ATM mutation in mCRPC. Attempts are being made to predict the larger patient population who can benefit from DDR inhibitors. Single-agent activity of olaparib has been reported in patients with sporadic cancers, which is likely explained by the presence of non-BRCA1/2 HR defects conferring susceptibility to PARP inhibition.32,33 Nevertheless, reliable biomarkers of response to PARP inhibition are yet to be defined and the increasing availability of genomic analysis can be expected to add meaningful information for patient selection.21,77
Several oncogenic features, such as alterations of replication timing and progression, lead to replication stress and are thus proposed as potential biomarkers for DDR inhibitor response, including ATR and ATM inhibitors. 78 RAS mutant cancers have been shown to have dependence on the DDR and KRAS mutations have been shown to induce hypersensitivity to ATR in cell lines, 79 as have CCNE1, CCND2 and MYC. 80 In ovarian cancer, lipid phosphatase inositol polyphosphate 4-phosphatase type II (INPP4B) loss (found in 40% of patients) causes a DNA repair deficit. 81 Other cell-cycle regulators including CDC25A also increase replication stress. 82
Aberrations in DDR/cell-cycle checkpoint genes cause replication stress in preclinical data. These include aberrations in including FA, Rb, 80 ERCC1, ribonucleotide reductase, 83 XRCC1 49 and ATM, 84 and can thus be proposed as potential biomarkers for DDR inhibition.
The SWI/SNF chromatin-remodelling complex is composed of multiple components including ARID1A, ARID1B, SMARCA4 and SMARCB1, and modulates DNA replication, transcription and repair.85,86 Defects in ARID1A sensitize tumour cells to ATR inhibition via topoisomerase 2A and cell-cycle defects. 87 Other epigenetic modulators including loss of the chromatin-remodelling protein ATRX and H3K36me3-deficiency have been shown to render cells hypersensitive to CHK1 and ATR inhibition.88,89
Significant heterogeneity characterizes DNA repair defects; prevalence varies across different tumour types. MSI occurs more frequently in colorectal cancer, whereas HR defects are more frequently detected in breast and ovarian cancers. 90 While genome sequencing has broadened the detection of DDR defects in other tumour types, precise estimates across different cancers are yet to be defined. In addition, the degree to which each DNA repair deficiency constitutes a catastrophic event, therefore rendering cells more susceptible to DDR inhibition, is still a point of uncertainty. Heterogeneity also exists in the impact of different DNA repair defects on patient outcome – for example, MSI colorectal cancers are characterized by better prognosis compared to genomically stable CRC. 91 These aspects constitute a limitation in the development of DDR agents. Despite encouraging results from early-phase trials, development is still challenged by lack of predictive biomarkers. A better understanding of each deficiency in the context of specific tumour types and the identification of validated biomarkers for patient selection will be critical for the development of these compounds.
Genomic scars
In DDR-deficient cells, DNA damages accumulates. This ‘genomic scar’ has different features depending on the pathway affected. For example, in HR deficiency there are large genomic deletions and loss of heterozygosity (LOH).92–94 BRCA1/2 mutations manifest as tandem duplications, and microhomology-mediated deletions. 95 Mismatch repair manifests as microsatellity instability. 95 The use of genomic scars as a predictive marker is not established. In the ARIEL3 trials of rucaparib maintenance following platinum chemotherapy in ovarian cancer, LOH was assessed. In those patients with BRCA wildtype tumours and LOH, 30% of patients in the rucaparib group achieved a benefit of over 1 year compared to 5% in the placebo group. LOH was, however, clearly not completely predictive of benefit. 96 In the NOVA trial of niraparib maintenance following platinum chemotherapy in ovarian cancer, HR deficiency was assessed by the myChoice HRD test which measures LOH, large-scale transitions and telomeric allelic imbalances. The HRD score did not predict niraparib benefit. 38
Functional assays
Functional assays are being developed, that aim to give a real-time read-out of DNA repair. These include the RAD51 focus formation assay that has been shown to correlate with HRR defects97,98 and the gamma-H2AX foci that correlates with DNA DSBs.99,100 Assays for replication stress include the DNA fibre assay. This technique utilizes labelled nucleotides that become incorporated into DNA at the activated replication forks. Cells are lysed and DNA fibres stained using fluoroprobes and visualized using fluorescent microscopy. Information or origin firing, elongating forks and replication fork stalling can thus be obtained.101,102 Clinical validation is awaited.
DDR inhibitors and the immune system
In recent years the key role that immune modulation has in oncogenesis has been recognized and the classic “hallmarks of cancer” have been updated to include “evasion of the immune system” as a key factor in tumourigenesis. 1 Emerging work is also revealing a tight coordination between the DDR and the immune defence systems (Figure 2).
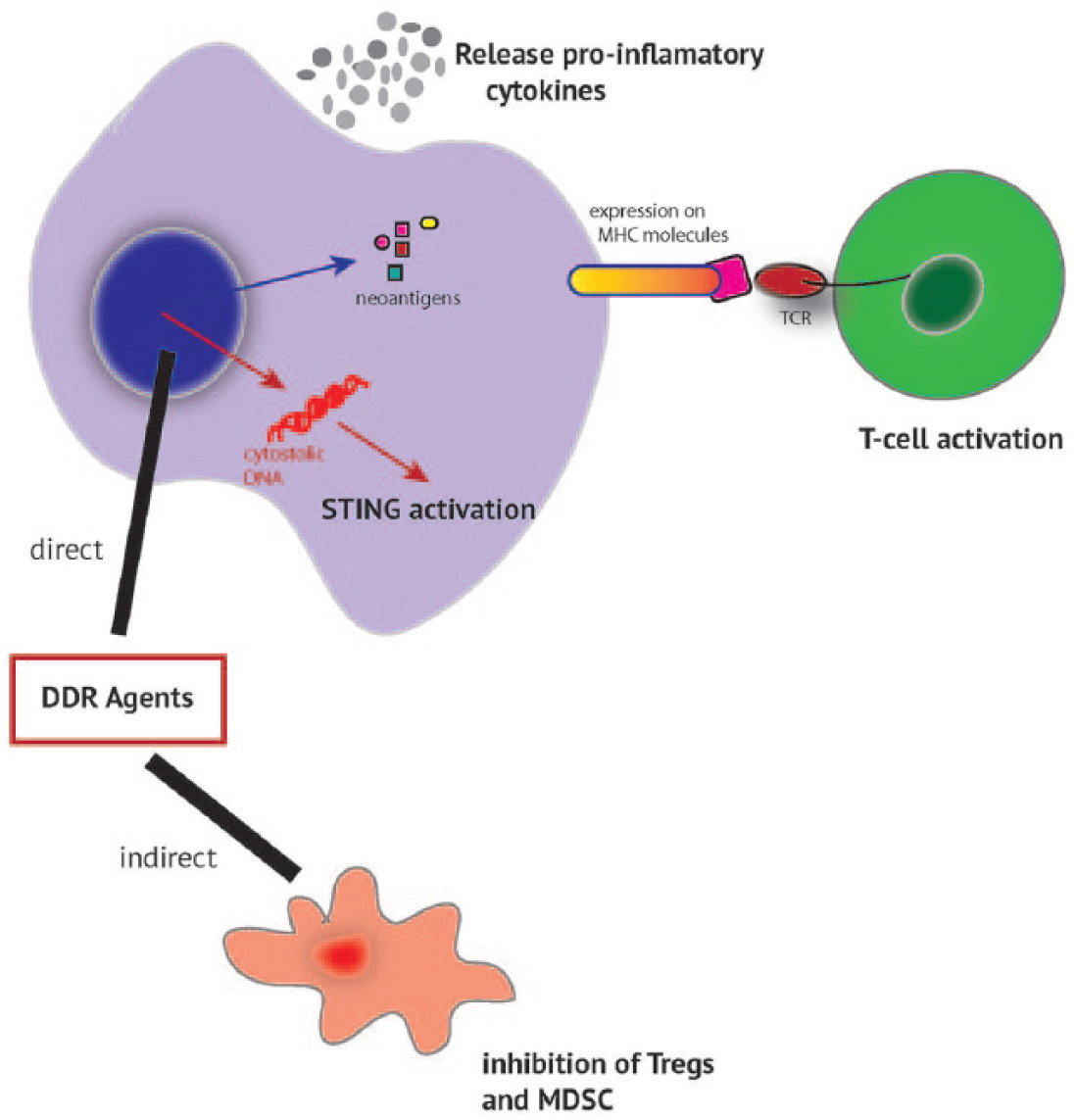
DDR agents and interaction with the immune environment.
Mutational burden and the immune system
DNA damage and repair influences responses to immunotherapy. 103 The burden of somatic mutations varies greatly between tumours, with melanoma, lung and bladder having the highest mutational load. 95 Mutations that cause a change in protein expression result in mutant proteins being bound by MHC class I and presented to T-cells, resulting in T-cell stimulation. These proteins are known as neoantigens and can arise from any changes that alter the open reading frame (ORF) sequences in the genome, such as missense mutations, fusion transcripts, frameshifts and stop losses. 104 Thus, neoantigen expression is closely correlated with mutational load.105–107
It has been found that tumour mutational load correlates with response and survival in CTLA-4 antagonists in metastatic melanoma.105,106 This has also been demonstrated in non-small cell lung cancer, where, in two independent cohorts, higher nonsynonymous mutation burden in tumours was associated with improved objective response, durable clinical benefit and PFS. 107 Tumours with a mutational landscape in which C > A transversions are common, typical of tobacco exposure, are more likely to benefit from immune checkpoint inhibition. 108 Measures of mutational load have classically been burdens of single nucleotide variants (SNVs). It has also been demonstrated that a number of small insertions and deletions (indels) cause frameshifts correlating with immunogenicity in a pan-cancer panel with correlation with immune checkpoint inhibitor responses seen in melanoma. 109
Neoantigen expression has been shown to correlate with response to immune checkpoint inhibitor and survival.105–107 Loss of neoantigens is also implicated in immune checkpoint inhibitor resistance. 110 The clonality of neoantigens is thought to play a role in response; loss of clonal expression of neoantigens is associated with immune checkpoint inhibitor resistance.110,111
DDR, mutational burden and response to immune checkpoint inhibitors
Deficits in DDR increase mutational burden; indeed, on large-scale genomic screens, defects in components of the DDR (including BRCA1/2 and ATM) result in unique mutational signatures in tumours.95,112 It can also be expected that the mutational burden will result in neoantigen burden and so influence responses to immunotherapy. Defects in DDR result in distinct immunological characteristics – for example, in breast cancer BRCA1/2-mutant tumours having higher levels of tumour-infiltrating lymphocytes and PD-L1.113,114
The most well-established example of DDR deficit and its influence on response to immunotherapy comes from MMR-deficient tumours. It has been demonstrated the MMR-deficient colorectal cancers have an activated immune microenvironment and upregulation of immune checkpoints such as PD-L1 and CTLA-4. 115 A phase II trial of the PD-1 inhibitor pembrolizumab demonstrated an objective response rate of 40% for MMR-deficient non-colorectal cancer patients and 71% for MMR-deficient colorectal cancer patients, contrasting to 0% for MMR-proficient patients. This clearly correlated with mutational load, with 1782 somatic mutations in MMR-deficient tumours compared to 73 somatic mutations in MMR-proficient tumours. 116 MMR-deficient tumours had a higher somatic mutational burden and neoantigen load. The study has since expanded to 12 MMR-deficient tumour types with objective radiographic responses seen in 53% of patients, and complete responses in 21% of patients. Responses were durable, with median PFS and overall survival not yet reached. 117 Following this trial, the FDA has approved pembrolizumab for treatment of MMR-deficient tumours.
In endometrial cancer with POLE mutations causing MSI there is a higher number of CD3+ and CD8+ tumour-infiltrating lymphocytes and increased PD-1 expression on tumour-infiltrating lymphocytes compared to microsatellite stable tumours. 118 In trials of pembrolizumab in non-small cell lung cancer, patients with prolonged responses were more likely to have mutations in DDR genes such as POLE, POLD1 and MSH2. 107 In melanoma patients treated with the PD-1 inhibitors pembrolizumab or nivolumab, a high proportion of responders (6/21) had a BRCA2 mutation compared to 1/17 non-responders. 119 Breast cancer patients with BRCA1/2-mutated tumours have a greater number of clonal mutations compared to wildtype. 120
DDR, immune cytokines and STING
DNA damage results in an increase in levels of inflammatory cytokines, including TNF-α and IL-6 via ATM and ATR.121,122 PARP inhibition synergizes with CTLA-4 blockade in a mouse ovarian model via IFN-gamma secretion. 123 The stimulator of interferon (STING) pathway plays a key role in innate immunity. Agonists of the STING pathway have been identified as enhancing anti-cancer immunity, with inhibitors of the pathway (β-catenin/wnt) inhibiting anti-cancer immunity. 124 STING pathway activating drugs are in development.125,126 DDR is intimately linked to the innate immune system. 127 There is evidence that cytosolic DNA sensors directly activate the STING pathway,128,129 which activate type I interferons which are known to augment cytotoxic T-cell priming, 130 and promote immunogenic cell death. DDR pathways may also directly activate the STING pathway.131,132
DDR and downregulation of MDSC and TREGs
Regulatory T-cells (TREGS) have an immunomodulatory role. DNA damage resulting from chemotherapy such as cyclophosphamide, temozolamide, gemcitabine and 5-FU has been shown to reduced TREG levels.133–136 Whether DDR inhibitors can indirectly cause a similar effect is, as yet, unknown.
DDR inhibitors in combination with immunotherapy
Given the interplay of DDR pathways and the immune system, synergy of DDR inhibitors and immunotherapy can be proposed. DDR inhibitors could increase the mutational burden, making tumours more immunogenic. However, it is, as yet, unknown whether treatment with a DDR inhibitor results in a similar change in mutational load, neoantigen profile or STING pathway activation as occurs with DDR genomic deficits. Some early preclinical data are supportive. BMN 673, a PARP inhibitor, increases CD8+ T-cells and increased IFN-gamma and TNF-α in BRCA1-deficient murine ovarian cancer, 137 and PARP inhibition has also been shown to upregulate PD-L1. 138 Clinical trials of combination immunotherapy and DDR inhibitors are ongoing (Table 2). 4
Conclusions and future challenges
We are at an exciting time in the clinical development of DDR inhibitors, where the dynamic interplay between DDR, therapeutic inhibition and the immune response offers a window of opportunity for the augmentation of anti-tumour effects.
For the patient-facing clinician, the take-home message is that a subset of cancers can be molecularly stratified for treatment with DDR inhibitor agents. While the frequency of DNA repair defects in each specific subtype of cancer remains to be determined, access to molecular profiling is becoming more widespread and affordable. The identification of patients with DDR defects should prompt early referrals for inclusion into clinical trials of rational combinations of DDR inhibitors which will hopefully lead to improved patient outcomes and survival (Figure 3). Envisaging the future, adaptive combinatorial treatments with DDR inhibitors tailored to match evolving tumour profiles and their resulting vulnerabilities is likely to become a reality.

The DDR treatment paradigm.