
Abstract
Despite decades of research, pancreatic ductal adenocarcinoma (PDAC) continues to have the worst 5-year survival of any malignancy. With 338,000 new cases diagnosed and over 300,000 deaths per year globally there is an urgent unmet need to improve the therapeutic options available.
Novel immunotherapies have shown promising results across multiple solid tumours, in a number of cases surpassing chemotherapy as a first-line therapeutic option. However, to date, trials of single-agent immunotherapies in PDAC have been disappointing and PDAC has been labelled as a nonimmunogenic cancer. This lack of response may in part be attributed to PDAC’s unique tumour microenvironment (TME), consisting of a dense fibrotic stroma and a scarcity of tumour infiltrating lymphocytes. However, as our understanding of the PDAC TME evolves, it is becoming apparent that the problem is not simply the immune system failing to recognize the cancer. There is a highly complex interplay between stromal signals, the immune system and tumour cells, at times possibly restraining tumour growth and at others supporting growth and metastasis.
Understanding this complexity will enable the development of rational combinations with immunotherapy, priming the TME to offer immunotherapy the best chance of success. This review seeks to describe the unique challenges of the PDAC TME, the potential opportunities it may afford and the trials in progress capitalizing on recent insights in this area.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is predicted to become the second leading cause of cancer-related deaths in America by 2030. 1 Despite a multitude of clinical trials, prognosis remains dismal, with a median overall survival (OS) of 4–6 months, having not significantly improved over the last 40 years. 2 This poor prognosis is multifactorial, attributed to PDAC’s systemic and aggressive nature, its complex mutational landscape, its desmoplastic stroma, a potently immunosuppressive tumour microenvironment (TME) and a current lack of effective therapies.
Surgery remains the only curative treatment for PDAC, but few patients present with operable disease and approximately 80% patients who undergo curative intent surgery ultimately relapse and succumb to their disease.3,4 The majority of patients present with advanced disease, where the standard of care is chemotherapy, but PDAC constitutes a relatively chemotherapy-resistant cancer. Even for the fittest patients, able to tolerate the triplet chemotherapy regimen FOLFIRINOX (5-fluorouracil, irinotecan and oxaliplatin), OS is only extended to 11 months. 5 Such palliative chemotherapy may be associated with significant toxicity and its impact on quality of life must be carefully considered. Targeted therapies in unselected PDAC patients have not fared any better than chemotherapy in clinical trials and have been unable to offer any clinically meaningful benefit to date. 6 There is therefore an urgent unmet need to develop novel, effective, well-tolerated treatments for this disease and immunotherapy is an obvious area for exploration.
Immunotherapy has resulted in a paradigm shift in the treatment of a number of solid tumours, including melanoma, non-small cell lung cancer (NSCLC), gastric cancer, genitourinary cancers, head and neck cancer and selected colorectal cancers. 7 However, as yet PDAC has proved more of a challenge with disappointing results from early trials of single-agent immune checkpoint blockade8,9 (Table 1). This failure is likely due to a combination of mechanisms of immune escape in PDAC. These mechanisms range from a potential lack of antigenicity and a low mutational burden to the complex interactions between tumour cells, the desmoplastic stroma and immune cells in PDAC creating a highly immunosuppressive TME, making this disease insusceptible to single-agent immunotherapy.
Single-agent trials of checkpoint inhibition in PDAC to date.

PDAC, pancreatic ductal adenocarcinoma; PD-L1, programmed death ligand 1.
This review seeks to describe PDAC’s immune escape mechanisms, focusing on recent insights into the interplay between various elements of its immune-excluded TME, and consider how these insights may be leveraged into combination immunotherapy studies with a sound scientific basis.
Antigenicity and tumour mutational burden in PDAC
As described in the cancer immunity cycle, an effective anticancer immune response requires multiple steps. 10 The first steps require the release and presentation of neoantigens: tumour-associated antigens or tumour-specific antigens. The presence of these neoantigens has been associated with increased numbers of tumour infiltrating lymphocytes (TILs) and enhanced sensitivity to checkpoint blockade. For example, a higher neoantigen burden and nonsynonymous mutational load have been associated with improved efficacy of pembrolizumab treatment in patients with NSCLC 11 and a higher mutational burden with increased clinical benefit from ipilimumab/tremelimumab in melanoma. 12 The relationship between high mutational burden, increased TILs and efficacy of immunotherapy is also seen in tumours associated with mismatch repair (MMR) deficiency, 13 noting approximately 9–17% of PDACs may have an MMR deficiency.14–16 While these microsatellite instability high PDAC patients may be considered for immunotherapy under the tumour agnostic approval for pembrolizumab, this is not the case for the majority of patients.
Pancreatic cancer is reported to have a relatively low mutational load with a median somatic mutational prevalence of only 1 mutation/megabase, contrasting with over 10 mutations/megabase present in melanoma and just under 10 for lung cancer and bladder cancer. 17 A value of 10 somatic mutations/megabase of DNA corresponds to approximately 150 nonsynonymous mutations within expressed genes and the formation of neoantigens is common in tumours with such a mutational load. 18 How effective neoantigen formation is with lower mutational loads of one or less is less clear. 18
Despite PDAC having a comparatively low mutational load there is evidence that nearly all cases do express some candidate neoantigens, including quality neoantigens predicted to have a robust level of expression on human leukocyte antigen class 1 molecules. 19 Further, using in silico neoantigen prediction, it has been demonstrated that patients with tumours with the highest number of neoantigens alongside the most abundant CD8+ T-cell infiltrates have the longest survival. 20 However, these neoantigens then require efficient presentation by antigen-presenting cells to stimulate a T-cell response, which appears to be problematic in PDAC.
Dendritic cells (DCs), a form of antigen-presenting cell, respond to neoantigen recognition with upregulation of the major histocompatibility complex (MHC) I and II and costimulatory molecules that interact with and activate T-cells. DCs in PDAC tend to be scarce and if present, immature, resulting in impaired early tumour antigen recognition and subsequent T-cell response. 21
In addition to the low mutational load and impaired antigen recognition, immunosuppression is also a particularly dominant force in PDAC, leading to actively suppressed T-cells with a reduced activation signature. 19 The TME plays a key role in this immunosuppression.
The tumour microenvironment in PDAC
The TME in PDAC is characterized by a desmoplastic reaction, a growth of fibrous tissue, surrounding the malignant epithelial cells. 22 This reaction is composed of cancer-associated fibroblasts, arising from pancreatic stellate cells, which produce several extracellular matrix proteins and cytokines, and vascular endothelial cells, all infiltrated by a variety of immune cells (lymphocytes, mast cells and macrophages; Figure 1).
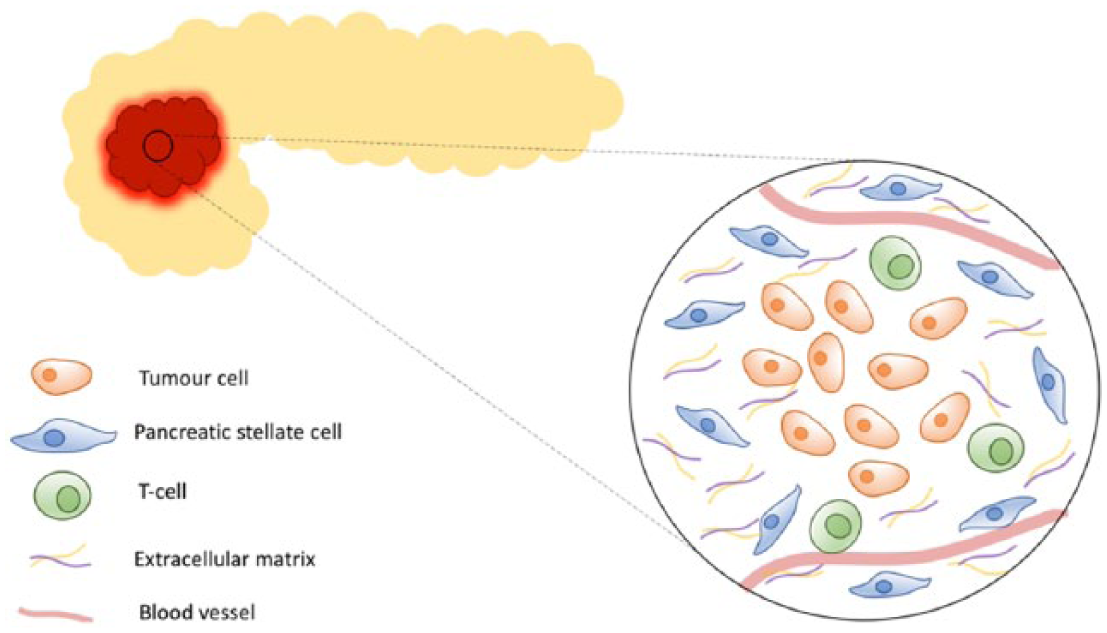
Pancreatic ductal adenocarcinoma stroma.
The highly fibrotic stroma is seen surrounding both primary and metastatic lesions 23 and is thought to play an important role in PDAC growth, metastasis and resistance to treatment, as well as promoting a hypoxic microenvironment. 24 Indeed, co-injection of human pancreatic stellate cells with tumour cells has been shown to result in increased primary tumour incidence, size, and metastasis in orthotopic mouse models. 25 However, there is considerable complexity to the interactions between the stroma and the tumour cells. The stroma appears to have a dual nature, at times even restraining pancreatic cancer progression.26–28
Moffitt and colleagues defined ‘normal’ and ‘activated’ stromal subtypes, based on stromal gene expression. The ‘activated’ stromal subtype is associated with a worse median OS versus the ‘normal’ stromal subtype [hazard ratio (HR) 1.94, confidence interval (CI) 1.11–3.37, p = 0.019]. 29 The group postulated that the existence of these two subtypes might help to explain the differential effects of stroma seen in some preclinical models and indeed in clinical trials. Activated stroma was characterized by a diverse set of genes associated with macrophages, such as ITGAM, an integrin and CCL13 and CCL18 chemokine ligands.
Unpicking such stromal signalling is of paramount importance in understanding how the immunosuppressive TME develops and is maintained. While PDAC has been described as a nonimmunogenic cancer a robust infiltrate of immune cells has been documented, usually dominated by myeloid derived suppressor cells (MDSCs), tumour-associated macrophages (TAMs) and neutrophils, with TILs present but in smaller numbers.19,30,31 The immunosuppressive MDSCs and TAMs are attracted to the TME by granulocyte macrophage colony-stimulating factor (GM-CSF) and chemokine (C-C motif) ligand 2 (CCL2) secreted by the tumour cells respectively. 32 The presence of these myeloid cells is associated with a worse prognosis in patients with resected disease, as are regulatory T-cells. On the other hand, the presence of effector (CD8+ and CD4+) T-cells may be associated with a favourable prognosis.33–36 The B-cells present are also thought to be important, with an interleukin (IL)35-producing CD1d(hi)CD5(+) subset demonstrated to accumulate in the TME during early neoplasia, supporting tumour cell growth. 37
Despite the presence of these immune cells and a theoretically inflamed TME, PDAC is still considered an immune-excluded tumour, meaning that while some TILs may be present they are prevented from directly interacting with the tumour cells, existing as clusters, tertiary lymphoid aggregates or trapped within the stroma.38,39 Those T-cells, which are present in the TME, may also not be able to mount a full immune response to the tumour cells, being hindered by the secretion of immunosuppressive cytokines such as IL-10 and transforming growth factor (TGF)-β, and inactivated by the loss of CD3 zeta, a signal transducing chain in TILs. 40 In addition, regulatory (FOXP3+) T-cells in the area block effector T-cell division and both macrophages and γδ T-cells, another type of immunosuppressive T-cell, prevent effector T-cells entering the TME through mechanisms including programmed cell death (PD)-1/programmed death ligand (PD-L)1 signalling. 41 Such escape mechanisms have been well documented in cancers with T-cell inflamed TMEs. 42
The type of T-cells present appear to be dynamic through the course of disease, with the prevalence of regulatory T-cells increasing from premalignant pancreatic lesions to advanced PDAC and certain chemotherapies such as gemcitabine and cyclophosphamide, able to transiently reduce the number of regulatory T-cells present.35,43,44 Understanding the type, location and functionality of the immune cells in the TME at different times in the disease process and during treatment will be particularly important when it comes to considering combination therapies to overcome the immunosuppressive tumour milieu.
Combinatorial strategies to overcome the immunosuppressive nature of PDAC
As described, there are multiple reasons why single-agent immunotherapy may fail in this tumour type with its low tumour antigenicity, poor presentation of neoantigens and a TME where the few T-cells present are prevented from interacting with the tumour and are suppressed by TAMs, MDSCs, regulatory T-cells and cytokines. As our understanding of this complex and challenging situation develops, novel combination strategies are being considered to target these various elements in an attempt to maximize the chance of success with immunotherapy.
Combination of chemotherapy with immunotherapy
Chemotherapies, including anthracyclines, gemcitabine and oxaliplatin, have been implicated in DC recruitment and activation.45,46 Induction chemotherapy may also trigger tumour-specific antigen release.47,48 This, and the transient reduction of regulatory T-cells seen with chemotherapies such as gemcitabine and cyclophosphamide, provides a sound rationale for using chemotherapy to prime the immune system, supporting the premise of a combined or possibly staggered chemotherapy and immunotherapy approach to treatment.
This strategy has been explored in a number of small early studies with some interesting results to date, noting these were mainly dose-escalation studies often in combination with single-agent gemcitabine (Table 2). In addition to the more commonly investigated checkpoint inhibitors such as CTLA-4 inhibitors (ipilimumab and tremelimumab) and anti-PD-1/PD-L1 antibodies (nivolumab, pembrolizumab and durvalumab) these trials also involve some novel immunotherapy approaches.
Selected chemotherapy and immunotherapy combination studies in PDAC.

AE, adverse event; BID, twice daily; CI, confidence interval; DCR, disease control rate; DLT, dose limiting toxicities; DoR, duration of response; GI, gastrointestinal; IDO, indoleamine-pyrrole 2,3-dioxygenase; IV, intravenous; MTD, maximum tolerated dose; OR, objective response; ORR, objective response rate; OS, overall survival; PD-1, programmed cell death 1; PDAC, pancreatic ductal adenocarcinoma; PD-L1, programmed death ligand 1; PFS, progression-free survival; PO, by mouth; BD, twice daily; PR, partial response; pts, patients; RP2D, recommended phase 2 dose; RR, response rate; TEAE, treatment emergent AEs; TTP, time to progression; TTR, time to response.
For example, targeting CD40 with an agonistic antibody aims to stimulate antigen-presenting cells, such as DCs and B-cells, and promote anti-tumour T-cell responses. 52 In PDAC mouse models, CD40 agonists have been demonstrated to enhance chemotherapy efficiency by redirecting TAMs to induce fibrosis degradation through interferon (IFN) and CCL2 signalling. 53 In a small clinical trial (n = 22 patients) the addition of a CD40 agonist to gemcitabine resulted in a median OS of 8.4 months and a response rate of 19%, which compared favourably against historical controls of single-agent gemcitabine, and further study is warranted. 50
C-C chemokine receptor type 2 (CCR2), a receptor for CCL2, is also a target of interest as it is involved in the recruitment of immunosuppressive TAMs into the PDAC TME. Colony-stimulating factor-1 receptor (CSF1R) provides another option, mediating the biological effects of CSF1, namely the production and differentiation of macrophages, including TAMs. Preclinical experiments have shown that inhibiting either CSF1R or CCR2 decreases the numbers of pancreatic tumour initiating cells and improves chemotherapeutic efficacy. 54 Further, CSF1R inhibition has been demonstrated to upregulate T-cell checkpoint molecules, including PDL1 and CTLA4 and be synergistic with checkpoint inhibition in PDAC models. 55 Following a phase I study which found the combination to be well tolerated and active in PDAC, trials of CCR2 inhibitors are ongoing with FOLFIRINOX, although interestingly a trial in combination with gemcitabine/Nab-paclitaxel was recently terminated [ClinicalTrials.gov identifier: NCT02732938]. CSF1R inhibitors are being studied in combination with other immunotherapies [ClinicalTrials.gov identifiers: NCT02526017/ NCT02777710/ NCT03153410].
Indoleamine-pyrrole 2,3-dioxygenase (IDO) is an enzyme implicated in the generation of an immunosuppressive TME through converting antigen-presenting cells from being immunogenic to tolerogenic, producing inhibitory cytokines and activating regulatory T-cells, and provides another focus of study. 56 In PDAC upregulation of IDO has been associated with an increased number of regulatory T-cells. 57 The interim analysis of the clinical trial of indoximod in combination with gemcitabine/Nab-paclitaxel listed below [ClinicalTrials.gov identifier: NCT02077881] reported a 37% response rate with one patient having a confirmed partial response 58 and the final results are awaited with interest.
Ibrutinib, a small molecule inhibitor of Bruton’s tyrosine kinase which blocks B-cell receptor signalling used in the treatment of various haematological malignancies, is also under investigation in PDAC. In mouse models of PDAC, ibrutinib limits tumour growth, diminishes fibrosis, extends survival, and improves the response to chemotherapy 59 and the results of a number of clinical studies are expected shortly.
It remains to be seen whether the future treatment of PDAC will involve immunotherapy in combination with chemotherapy and its attendant toxicities. At the moment, while these novel immune targets are being assessed, a chemotherapy–immunotherapy combination appears to be a judicious approach for clinical trials. This is especially true with the newer combination chemotherapy regimens with a reasonable response rate, which can be important in patients with bulky disease. Once the most promising targets are selected it will be interesting to see if chemotherapy free treatment options become a reality.
Vaccine combinations
A multitude of vaccines have been studied in PDAC including whole-cell, DC, specific peptide and virus-based vaccines. Multiple antigen targets have been investigated with mesothelin, mucin-1 (MUC1), Wilms’ tumour 1 (WT1), carcinoembryonic antigen (CEA) and mutated KRAS making some of the most appealing targets60,61 (Table 3). While a couple of small studies have demonstrated that personalized peptide vaccination in combination with chemotherapy may be a well-tolerated and potentially interesting approach in this disease, we are some way away from this becoming a practical therapy.62,63
Selected antigen targets in PDAC.

CEA, carcinoembryonic antigen; PDAC, pancreatic ductal adenocarcinoma.
One of the most studied vaccines to date is GVAX, a whole-cell vaccine. Whole-cell vaccines enable multiple antigens to be targeted simultaneously and as such may result in an expanded T-cell repertoire. Such vaccines may be derived from a specific patient’s tumour (autologous vaccines), or from another patient’s tumour (allogeneic vaccines). Allogeneic whole-cell vaccines appear to provide a more pragmatic approach and multiple studies have been conducted using the GVAX vaccine in a variety of settings with some interesting if mixed results.
GVAX is an irradiated whole-cell tumour vaccine which has been genetically modified to release GM-CSF, a cytokine that mobilizes leukocytes to the TME and induces significant immunoglobulin (Ig)G and IgM responses. 73 GVAX has been shown to induce T-cell infiltration and the formation of tertiary lymphoid aggregates in patients with PDAC, when administered prior to resection, possibly converting a ‘nonimmunogenic’ tumour into a more ‘immunogenic’ tumour type. 74
GVAX therapy has also been associated with a significant upregulation of PD-L1 expression in PDAC mice models. When combined with an anti-PD-1 antibody, the mice were found to have increased CD8+ T-cells in the TME and associated improved survival compared with either treatment alone. 75 These findings are supported in vivo with Lutz and colleagues describing upregulation of the PD-1/PD-L1 pathway in PDAC patients treated with GVAX. 74 Immune priming with GVAX may therefore improve clinical response to checkpoint inhibition immunotherapy. A phase Ib trial of 30 PDAC patients investigated the anti-CTLA-4 antibody ipilimumab alone or combined with GVAX, with an improved 1-year OS of 27% versus 7% and OS of 5.7 versus 3.6 months [hazard ration (HR) = 0.51, p = 0.072] in the combination group. 76 This study also demonstrated an enhanced T-cell repertoire and increase in peak mesothelin-specific T-cells, suggesting an immune-primed state with combination GVAX and ipilimumab. Various studies are underway investigating GVAX with a checkpoint blockade (Table 4.).
Selected ongoing vaccine/immunotherapy combination studies in PDAC.

PDAC, pancreatic ductal adenocarcinoma; SBRT,
GVAX has further been studied in combination with CRS-207 (an attenuated strain of Listeria monocytogenes engineered to express mesothelin) following intriguing preclinical studies. These mouse studies demonstrated the depletion of regulatory T-cells and a significantly improved survival (OS of 265 versus 150 days) in early pancreatic intraepithelial neoplasms following vaccination with Listeria monocytogenes in combination with an anti-CD25 antibody and cyclophosphamide. The combination was found to increase immunostimulatory IL-17 and IFNγ-secreting CD4+ T-cells. 77
A phase II study (n = 90) comparing GVAX/cyclophosphamide alone or followed by CRS-207 in previously treated metastatic PDAC demonstrated an OS of 9.7 versus 4.6 months (HR 0.53, p = 0.02) in favour of the combination arm. 78 Prolonged OS was associated with an enhanced mesothelin-specific CD8+ T-cell response in both arms. However, a larger randomized phase IIb study (n = 303), ECLIPSE, of GVAX/cyclophosphamide/CRS-207 versus CRS-207 alone versus chemotherapy failed to meet its primary endpoint of improving OS, according to a press release by Aduro Biotech. Here, OS in this third or subsequent-line setting was 3.8 months for patients treated with the CRS-207/GVAX combination, 5.4 months for patients treated with CRS-207 alone and 4.6 months for patients treated with chemotherapy. The results of an additional study of GVAX/CRS-207 with or without nivolumab in the second or subsequent-line treatment of advanced PDAC are awaited to ascertain if combination with a checkpoint inhibitor fares any better [ClinicalTrials.gov identifier: NCT02243371, STELLAR study].
Algenpantucel-L is another allogenic whole-cell vaccine, engineered to express alpha-Gal (mouse alpha-1, 3-galactosyltransferase gene) in two human PDAC cell lines. Here recent trial results have also been less than encouraging, despite earlier positive results, with the phase III IMPRESS study failing to reach its primary endpoint of improving OS. A press release by New Link genetics confirmed OS for patients with respectable PDAC treated with surgery, standard of care and adjuvant algenpantucel was 27.3 months versus 30.4 months for those treated with surgery and standard of care alone.
The varied results from preclinical and clinical vaccine studies suggest that, while some vaccines may be active in this disease, it is unlikely that a single-agent vaccine approach will be able to successfully overcome the level of immunosuppression seen in PDAC. The results of the various ongoing checkpoint inhibitor/vaccine combination studies are awaited with interest (Table 4).
Adoptive T-cell strategies
Adoptive T-cell strategies, or cellular adoptive immunotherapy, is an approach whereby tumour reactive T-cells are collected, modified ex vivo and infused to generate an optimized immune response, most extensively investigated in haematological cancers. The T-cells may be derived from an endogenous source, autologous or allogenic cytotoxic T lymphocytes (CTLs), or be engineered to recognize a specific tumour antigen via a chimeric antigen receptor (CAR-T-cell) or a cloned T-cell receptor.
A number of preclinical and small clinical trials of a CTL infusion have been completed in PDAC. For example, MUC1-reactive CTLs, generated by exposing T-cells from healthy volunteers’ peripheral blood samples to a MUC1-expressing human PDAC cell line, have been shown to be cytotoxic against MUC1-expressing PDAC cell lines. 79 In a clinical study of CTLs in combination with pulsed MUC1 DCs, 5/20 patients with unresectable or recurrent PDAC had stable disease and 1 patient with multiple lung metastases had a complete response with a mean OS of 9.8 months and no grade 2–4 toxicity reported. 80 A further retrospective study investigated the outcomes for patients with unresectable or recurrent PDAC treated with MUC1-DCs, MUC1-CTLs and gemcitabine in combination. In the 42 patients analyzed, median survival was 13.9 months with a disease control rate of over 60% with no severe toxicities reported. 81 Further prospective randomized study appears to be warranted.
MUC1-targeting CAR-T-cells have also been investigated. In a PDAC xenograft model CAR-T-cells engineered to recognize the tumour-specific Tn glycoform of MUC1, a neoantigen, demonstrated target-specific activity, controlled tumour growth and improved survival. 82 A phase I/II clinical study in patients with MUC1-positive advanced solid tumours, including PDAC, is currently recruiting [ClinicalTrials.gov identifier: NCT 02587689]. CAR natural killer cells targeting MUC1 are also being studied in a similar patient population, including PDAC [ClinicalTrials.gov identifier: NCT02839954]. Other targets for CAR-T-cells under investigation in PDAC include mesothelin and CEA [ClinicalTrials.gov identifiers: NCT02465983, NCT02349724].83,84
While adoptive T-cell strategies provide a novel and exciting approach, there are many hurdles to overcome before this treatment reaches the clinic. Both infused TILs and CAR-T-cells have been shown to become progressively dysfunctional over time and to upregulate various inhibitory receptors including PD-1 and LAG3. 83 Further, depending upon the antigen selected for CAR-T therapy there is a risk of low level expression on normal tissues and the development of toxicity and autoimmunity, in addition to the risk of cytokine release syndrome. As with the other combination approaches discussed herein, choosing the correct partner for CAR-T therapy, as well as the most effective and safest antigen, will be of paramount importance.
Combination of agents targeting the stroma and immunotherapy
As discussed, the TME plays a critical role in PDAC and much effort has been spent in developing therapies to target its desmoplastic stroma. While some have been disappointing, notably the hedgehog inhibitors, 85 a number of more recent studies have been more promising and based on these results future combinations of agents aiming to remodel or reprogram the stroma and immunotherapy appear likely.
Hyaluronic acid (HA) is a large glycosaminoglycan, abundant in the PDAC extracellular matrix and correlated with a poor prognosis. 86 Following mouse studies demonstrating low vascularity and high interstitial pressure associated with high HA expression responding to treatment with hyaluronidase, clinical studies of PEGPH20, a pegylated recombinant human hyaluronidase, commenced.87,88 The latest to report is the randomized phase II HALO 202 study of PEGPH20 in combination with gemcitabine and nab-paclitaxel as a first-line treatment for metastatic PDAC versus gemcitabine and nab-paclitaxel alone. 89 A total of 34% of patients were found to be HA-high (defined as over 50% HA tumour surface staining). Progression-free survival was increased in the triplet regimen in all patients, but the largest improvement was seen in the HA-high patients, with an objective response of 45% versus 31%, and an OS of 11.5 versus 8.5 months (HR, 0.96; 95% CI, 0.57–1.61). Thromboembolic events were significantly increased in the triplet arm such that the study was put on hold and, in a second phase of the study, prophylactic enoxaparin was added. Following this amendment, the combination had a manageable toxicity profile, with thromboembolic event frequency reduced and no increase in bleeding, and a phase III study is underway [ClinicalTrials.gov identifier: NCT02715804].
Preclinical in vitro and in vivo studies have demonstrated that the barrier formed by high levels of HA in the TME, inhibits access of monoclonal antibodies and natural killer cells, and that combination therapy with PEGPH20 may enhance the anti-tumour effects of the monoclonal antibodies. 90 Further, tumour growth inhibition by anti-PD-L1 and anti-PD-1 drugs has been found to be enhanced by PEGPH20 in mouse HA-high PDAC models. 91 A phase Ib study of PEGPH20 in combination with pembrolizumab is underway in NSCLC and gastric cancer [ClinicalTrials.gov identifier: NCT02563548] and a phase I dose-escalation study of VCN-01, a genetically modified human adenovirus encoding human PH20 hyaluronidase, alone or in combination with gemcitabine/nab-paclitaxel is recruiting patients with advanced solid tumours, including PDAC [ClinicalTrials.gov identifier: NCT02045602].
Focal adhesion kinase (FAK), a nonreceptor cytoplasmic tyrosine kinase, provides another stromal target. FAK promotes tumour progression and metastasis through its effects both on cancer cells and on the stromal cells of the TME, where FAK phosphorylation aids epithelial to mesenchymal transition. Through kinase dependent and independent processes FAK integrates signals from integrins and growth factor receptors to regulate cell proliferation and survival, to promote angiogenesis, migration, invasion and cancer stem cell (CSC) renewal and its expression has been demonstrated in pancreatic cell lines and resected PDAC, where expression was correlated with tumour size and stage.92–94 While no clinical trials have yet demonstrated a response to single-agent FAK inhibition in PDAC, a synergistic effect was demonstrated preclinically when FAK inhibition was combined with chemotherapy and a PD-1 antagonist and a phase I study of this combination is underway [ClinicalTrials.gov identifier: NCT02546531, gemcitabine, defactinib and pembrolizumab].95–97
While the PDAC stroma clearly plays an important role in restricting the access of various therapies to the tumour, it is also thought to restrain tumour invasion and metastasis and novel targets have been sought to reprogram rather than ablate the stroma. The C-X-C motif chemokine receptor type 4 (CXCR4)/stromal derived factor-1 (CXCL12) axis provides such a target. It is thought to be important in driving invasion and metastasis in PDAC, with CXCR4 strongly expressed at the tumour’s leading edge in CSCs 98 and CXCL12 secreted by cancer-associated fibroblasts. This role may be mediated in part through CXCR4/CXCL12 activation of the Wnt/β-catenin axis and nuclear factor (NF)-κB which results in increased matrix metalloprotein secretion and a resulting decomposition of the extracellular matrix, enabling invasion. 99
In a preclinical study, CXCR4+ CSCs have been shown to be required for the development of liver metastases and the blockade of CXCR4 was found to significantly reduce metastasis in orthotopic mouse models of PDAC. 98 In addition, this axis has been implicated in mediating immunosuppression by cancer-associated fibroblasts and its inhibition with AMD3100 has been shown to act synergistically with an anti-PD-L1 therapy in a PDAC mouse model. 100 A number of early-phase clinical studies are underway testing the combination of a CXCR4 antagonist and a checkpoint inhibitor such as COMBAT, a phase II study assessing the combination of BL-8040 and pembrolizumab in patients with metastatic PDAC [ClinicalTrials.gov identifier: NCT02826486]. Although the CXCessoR4 phase I/II study of the anti-CXCR4, ulocuplumab, and nivolumab in PDAC and small cell lung cancer (SCLC) was terminated early due to a lack of efficacy [ClinicalTrials.gov identifier: NCT02472977] and the success of this approach remains to be seen.
Another stromal target thought to play an important role in invasion and metastasis is retinoic acid. In PDAC, quiescent pancreatic stellate cells transform into activated cancer-associated fibroblasts secreting extracellular matrix, remodelling and stiffening the TME. 101 All-trans retinoic acid (ATRA), a physiologically active form of vitamin A and retinoic acid receptors are reduced in PDAC tissue and associated with worse patient survival outcomes. 102 It has been demonstrated that ATRA can be used to restore mechanical quiescence and reduce the motility of pancreatic stellate cells, suppress extracellular matrix remodelling to inhibit invasion, reduce proliferation and increase cancer cell apoptosis in three-dimensional (3D) organotypic and mouse PDAC models.101,103 The STARPAC clinical study is in progress looking at the combination of ATRA, gemcitabine and nab-paclitaxel. Given the contributions of retinoic acid to immunological tolerance and the elicitation of adaptive immune responses, should this approach prove active, future combinations with checkpoint blockade may prove interesting.104,105
Radiotherapy combinations
Historically, radiotherapy had been considered to compromise the immune system, as white blood cells are highly sensitive to irradiation and the large fields delivered often caused damage to local lymphatics. However, with more modern highly localized techniques and a greater understanding of radiotherapy’s immunomodulatory and abscopal effects, where a patient may show disease regression at a site distant to the irradiated area, the role of radiotherapy as an immune priming treatment is now being explored across multiple solid tumours. 106
In a preclinical PDAC study, checkpoint inhibition with a PD-L1 inhibitor significantly improved tumour response to high dose radiotherapy by altering the phenotype of the TME to be more ‘anti-tumorigenic’. 107 In this study anti-PD-L1 therapy alone and in combination with radiotherapy significantly increased the CD8+ve/Treg ratio and enhanced the effect of radiotherapy preventing the formation of liver metastases. Further, Azad and colleagues demonstrated that PD-L1 inhibition also improved tumour response after gemcitabine based chemoradiation in a PDAC mouse model.
Following such interesting preclinical data, a number of clinical trials are now underway in this area. The IMPACT 2010 study aims to use low dose radiation to improve T-cell infiltration in resectable PDAC [ClinicalTrials.gov identifier: NCT01027221]. A clinical trial of immune checkpoint inhibition with radiotherapy in unresectable nonmetastatic PDAC will investigate the combination of durvalumab, tremelimumab or both with stereotactic body radiation therapy (SBRT) [ClinicalTrials.gov identifier: NCT02868632] while CheckPAC is investigating the combination of ipilimumab and nivolumab with radiotherapy in metastatic PDAC [ClinicalTrials.gov identifier: NCT02866383].
Other novel targets are being considered for use in combination with radiotherapy. Stimulator of interferon genes (STING) is a transmembrane protein implicated in the production of type 1 interferons and inflammatory cytokines in response to viral infections which also appears to play a role in the adaptive immune response against cancer.108,109 In a mouse model of PDAC, STING ligands have been shown to synergize with computed tomography (CT)-guided radiotherapy to control local and distant tumours through early tumour necrosis factor (TNF) α-dependent necrosis followed by later CD8+ T-cell-dependent control of remaining disease. 110 The authors suggest that the STING ligand converts cell death mediated by radiotherapy into an endogenous vaccine, enhancing the adaptive immune response, controlling local and distant disease. STING is expressed by human PDAC and stromal cells and it will be interesting to see if these results translate into positive clinical trials either in combination with radiotherapy or with checkpoint blockade.
In a similar vein, Toll-like receptors (TLRs), transmembrane proteins which play an important role in tissue repair and injury induced inflammation, may provide another novel target in combination with radiotherapy. In cancer, TLR agonists are thought to upregulate the adaptive immune response, induce vascular permeability and recruit leukocytes to the TME but have also been associated with promoting cancer survival and progression. 111 TLRs 7/8 are highly expressed in human PDAC and TLR 7/8 agonists have been shown to boost DC antigen-presenting activity, as an adjuvant to radiotherapy in mouse models of PDAC. 112 However, expression and stimulation of TLRs 7/8 have also been associated with cancer progression and resistance to fluorouracil (5-FU) in cell lines, possibly through Notch-2 signalling. 113 Early clinical trials of various TLR agonists are underway in combination with chemotherapy, immunotherapy and radiotherapy in advanced solid tumours, including PDAC (ClinicalTrials.gov identifier: NCT02650635, TLR8 agonist VTX-2337 and cyclophosphamide; ClinicalTrials.gov identifier: NCT02643303, tremelimumab, durvalumab and PolylCLC a TLR3 agonist; and ClinicalTrials.gov identifier: NCT03322384, IDO inhibitor epacadostat, TLR9 agonist SD101 and radiotherapy). The results of all of these studies are awaited with interest.
In addition to the potential of radiotherapy in combination with immunotherapy in PDAC, other local treatments such as radiofrequency ablation (RFA) or irreversible electroporation may also be used to prime the immune system. A small recent study of 10 patients with locally advanced PDAC undergoing RFA demonstrated a general activation of the adaptive immune response and a decrease in immunosuppression, lasting some weeks after the procedure. 114
Immunotherapy combinations
Single-agent checkpoint blockade has failed in this disease but various immunotherapy–immunotherapy combinations are under investigation. Dual checkpoint blockade has proved a popular approach and a number of studies are due to report shortly. The ALPs study of durvalumab ± tremelimumab in the second line treatment of metastatic PDAC has completed recruitment (n = 65) and the results are awaited [ClinicalTrials.gov identifier: NCT02558894].
The CheckMate032 study [ClinicalTrials.gov identifier: NCT01928394] is investigating nivolumab alone or in combination with ipilimumab across multiple solid tumours including PDAC. Interestingly, since Jan 2017, while the original nivolumab/ipilimumab arm in PDAC has not been expanded, an additional arm investigating the combination of nivolumab and ipilimumab with the MEK inhibitor cobimetanib has been introduced. Recent mouse colorectal cancer (CRC) studies have demonstrated that MEK inhibition in combination with anti-PDL1 therapy resulted in a synergistic and durable tumour regression attributed to a MEK inhibition dependent increase of CD8+ T-cells within the tumour. 115 Early data from the CRC cohort from the phase Ib study of cobimetinib and atezolizumab further supports this combination, demonstrating that microsatellite-stable (MSS) CRC may respond to anti-PDL1 therapy in combination with MEK inhibition and that the combination is tolerable [ClinicalTrials.gov identifier: NCT01988896]. 116 As MSS CRC, like PDAC, is considered an immune-insensitive cancer, these results may be particularly relevant.
Many other novel immune targets are also being considered in combination with checkpoint blockade, such as signalling via the C-X-C chemokine receptor type 2 (CXCR2) axis or IL10. The CXCR2 axis is an inflammatory signalling pathway involved in neutrophil recruitment, migration and tumour cell proliferation. In human PDAC CXCR2 signalling at the tumour border has been associated with a poor outcome.117,118 Interest in CXCR2/checkpoint inhibition was piqued by a mouse study where CXCR2 inhibition was demonstrated to promote T-cell tumour infiltration and increased sensitivity to anti-PD-1 immunotherapy. 119 A phase Ib/II trial is currently evaluating durvalumab in combination with either chemotherapy (nab-paclitaxel and gemcitabine) or CXCR2 inhibitor (AZD5069) in metastatic PDAC [ClinicalTrials.gov identifier: NCT02583477].
IL-10 has been considered to be an anti-inflammatory, protumourigenic cytokine, mainly secreted by M2-macrophages, regulatory T-cells and T helper 2 cells, with elevated levels of circulating IL-10 associated with a poor outcome in various cancers. 120 However, additional studies have suggested an anti-tumour role for IL-10, with IL-10 able to boost anti-tumour immunity in mouse studies expanding CD8+ TILs and inhibiting inflammatory CD4+ T-cells. 121 The phase I study of AM0010, pegylated recombinant human IL-10, suggests that IL-10 can act as an immune activating cytokine in human solid tumours, leading to systemic immune activation with increased immune-stimulatory cytokines and reduced TGFβ in patients’ serum. 121 In PDAC, AM0010 has been investigated alone and in combination with chemotherapy, demonstrating clinical activity and immune stimulation, with AM0010 increasing PD-1+ activated CD8 T-cells and stimulating an oligoclonal expansion of T-cell clones in the blood. 122 A phase I dose-escalation trial is currently underway with PDAC arms investigating the combination of daily AM0010 with chemotherapy or anti-PD-1 pembrolizumab or nivolumab [ClinicalTrials.gov identifier: NCT02009449].
Still more therapies targeting other novel checkpoints are in early development, including drugs directed at lymphocyte activation gene 3 (LAG3), T-cell immunoglobulin and mucin-domain-containing-3 (TIM3), T-cell immunoglobulin and immune-tyrosine inhibitory motif domain (TIGIT) and glucocorticoid-induced TNFR-related protein. Strategies to harness costimulatory molecules are also being considered, for example targeting CD137 or OX-40, both members of the TNF superfamily.123,124 A number of these targets are already being assessed in phase I studies, including PDAC patients. Once the optimal targets have been selected and potentially combined with other immunotherapies in a rational manner, it will be interesting to see if such combinations are sufficient to augment anti-tumour immunity without the need for chemotherapy in this most immunosuppressive disease.
An alternative approach to combination immunotherapy is the development of single drugs which are able to target more than one epitope. Bi-specifics and multi-specifics are antibodies which are engineered to have such multi-functionality and in cancer have been designed to block particular pathways more completely or to deliver effector immune cells efficiently to tumours. 125 For example, a phase I study of epidermal growth factor receptor (EGFR) bispecific antibody armed T-cells (BATs), anti-CD3 × anti-EGFR BATs, demonstrated clinical activity with a median OS of 14.5 months in five patients with locally advanced or metastatic PDAC and a phase Ib/II in the maintenance setting is ongoing [ClinicalTrials.gov identifier: NCT03269526]. 126 This field is very much in its infancy but early results are encouraging.
Combinations and DNA damage repair pathways
There is accumulating evidence that continued DNA damage in tumour cells results in a proinflammatory, immunologically active tumour environment.127,128 This effect may be heightened in tumours with deficient DNA damage repair and indeed such tumours have been demonstrated to be more sensitive to immunotherapy, as exemplified by the sensitivity of MMR-deficient colorectal cancer to checkpoint blockade versus MMR-proficient colorectal cancer. 13
Approximately 15% of PDAC patients fall into an ‘unstable’ molecular subtype, which is associated with deficiencies in DNA maintenance and a sensitivity to platinum agents. 129 Such features may be used to select patients for immunotherapy in the future. This approach is being investigated in a phase II study of the IDO inhibitor epacadostat in combination with pembrolizumab in PDAC patients with chromosomal instability or homologous recombination deficiency [ClinicalTrials.gov identifier: NCT03432676].
DNA repair deficiencies may also be used as a target themselves. Various poly ADP ribose polymerase (PARP) inhibitors have been investigated in PDAC, alone and in combination with chemotherapy, with some promising results.130–132 Further studies combining immunotherapy with PARP inhibitors are underway, such as Parpvax, a phase Ib/II study of niraparib plus either ipilimumab or nivolumab in patients with advanced PDAC whose disease has not progressed on platinum-based therapy [ClinicalTrials.gov identifier: NCT03404960].
Conclusion
PDAC presents an extremely difficult malignancy to treat. Its poor immunogenicity, unique TME and high levels of immunosuppression provide significant challenges when considering immunotherapy as a therapeutic option. However, as our depth of understanding increases, methods to overcome these hurdles are presenting themselves and a multiplicity of immunotherapy studies in PDAC are underway, considering innovative targets and scientifically sound combinations. Appropriate patient selection for these novel combination approaches will be of paramount importance and advances in molecular subtyping in PDAC may also be significant.14,133 Overall, with continued progress in understanding the immunobiology of this disease, there are reasons to be optimistic that immunotherapy may well play an important role in the treatment of PDAC in the future.
Footnotes
Acknowledgements
K. Young and D. J. Hughes are joint first authors. K. Young, D. Hughes and N. Starling contributed equally to this work; K. Young and D. J. Hughes reviewed the literature; K. Young, D. J. Hughes, D. Cunningham and N. Starling drafted the manuscript and made critical revisions prior to the final approval of this article.
Funding
The authors received funding from the National Institute for Health Research Biomedical Research Centre at the Royal Marsden NHS Foundation Trust and the Institute of Cancer Research.
Conflict of interest statement
K. Young and D. J. Hughes have no conflicting interests to declare, commercial, intellectual or otherwise. N Starling has received research funding from AstraZeneca, Bristol Myers Squibb, Merck; travel and accommodation expenses from AstraZeneca, Bristol Myers Squibb, Eli Lilly; honoraria from AstraZeneca, Eli Lilly. D. Cunningham has received research funding from Amgen, Sanofi, Merrimack, AstraZeneca, Celgene, MedImmune, Bayer, 4SC, Clovis, Eli Lilly, Janssen and Merck