
Abstract
Purpose:
Regorafenib monotherapy, a multikinase inhibitor of angiogenesis, tumor microenvironment, and tumorigenesis, showed promising results in gastric cancer. We aimed to assess the tolerability of regorafenib and paclitaxel in patients with advanced esophagogastric cancer (EGC) refractory to first-line treatment, and explore potential biomarkers.
Methods:
Patients received paclitaxel (80 mg/m2) on days 1, 8, and 15 of a 28-day cycle and regorafenib (80/120/160 mg) on days 1–21 in the dose-escalation cohort, and the maximum-tolerated dose (MTD) in the dose-expansion cohort. Exploratory, overall survival (OS) and progression-free survival (PFS) were compared to a propensity-score matched cohort receiving standard second-/third-line systemic treatment. Paclitaxel pharmacokinetics were assessed using samples from day 1 (D1) and day 15 (D15). We performed enzyme-linked immunosorbent assay measurements of galectin-1, RNA sequencing, and shallow whole-genome sequencing of metastatic tumor biopsies for biomarker analyses.
Results:
In the dose-escalation cohort (n = 14), the MTD of regorafenib was 120 mg. In all, 34 patients were enrolled in the dose-expansion cohort. Most common toxicities (all grades; grade ⩾ 3) were fatigue (79%; 4%) and sensory neuropathy (63%; 4%). Best responses achieved were partial response (28%) and stable disease (54%). Median OS and PFS were 7.8 and 4.2 months, respectively (median follow-up: 7.8 months). OS (p = 0.08) and PFS (p = 0.81) were not significantly improved compared to the matched cohort. Paclitaxel concentrations were significantly increased with regorafenib (D15) compared with paclitaxel only (D1; p < 0.05); no associations were observed with toxicity or efficacy. An increase in circulating galectin-1 compared to baseline was associated with shorter OS (p < 0.01). Enrichment of angiogenesis-related gene expression was observed in short survivors measured by RNA sequencing. Chromosome 19q13.12-q13.2 amplification was associated with shorter OS (p = 0.02) and PFS (p = 0.02).
Conclusion:
Treatment with regorafenib and paclitaxel is tolerable and shows promising efficacy in advanced EGC refractory to first-line treatment. Galectin-1 and chromosome 19q13.12-q13.2 amplification could serve as negative predictive biomarkers for treatment response.
Registration:
Clinicaltrials.gov, NCT02406170, https://clinicaltrials.gov/ct2/show/NCT02406170
Keywords
Introduction
Esophagogastric cancer (EGC) accounts for approximately 15% of all cancer deaths worldwide. 1 The majority of patients present with advanced disease and a median survival of less than 1 year. 2 First-line chemotherapy has demonstrated benefit over best supportive care while maintaining the patients’ quality of life.3 –6 However, the majority of patients do not achieve response or demonstrate progression of disease within a few months of first-line treatment. 2 As the gain in survival from second-line treatment is modest, there is an unmet need to improve beyond first-line treatment. 7
In the pathogenesis of EGC, angiogenesis has been identified as a key feature associated with adverse outcomes. 8 Regorafenib is an oral small-molecule tyrosine-kinase inhibitor, simultaneously targeting angiogenesis, tumorigenesis, and the tumor microenvironment. By targeting multiple membrane-bound and intracellular kinases, regorafenib has demonstrated anti-angiogenic potential, as well as inhibition of tumor growth and targeting of the tumor stroma in vivo. 9 In several solid tumors refractory to prior treatment, including gastric cancer, regorafenib monotherapy has demonstrated a survival benefit, as well as increased response rates compared to best supportive care only.9 –14 Similarly, efficacy of angiogenesis inhibition has previously been demonstrated in second-line advanced EGC using ramucirumab monotherapy, a monoclonal antibody against vascular endothelial growth factor (VEGF) receptor-2. 15 It has been suggested that the combination of paclitaxel and ramucirumab shows superior effects compared to monotherapy. 16 Although no direct comparison between paclitaxel with ramucirumab compared to ramucirumab monotherapy has been made, overall survival (OS) in the RAINBOW study suggests that the combination of angiogenesis inhibition and paclitaxel may be beneficial. 17 As the absolute gain in survival from ramucirumab with paclitaxel was still limited, we hypothesized that the combination of paclitaxel and regorafenib could result in increased efficacy compared to paclitaxel and ramucirumab, as the tyrosine-kinase inhibitor regorafenib not only targets angiogenesis, but also targets tumorigenesis and the tumor microenvironment. 16
Although previous studies demonstrated safety and response from combining regorafenib with a fluoropyrimidine and platinum-based compound in refractory metastatic colorectal cancer, 18 overall severe toxicities have been described for the combination of tyrosine-kinase inhibitors with cytotoxic chemotherapies.19 –21 Therefore, we first performed a phase I study with a dose-escalation cohort to assess the toxicity and the maximum-tolerated dose (MTD) of regorafenib. 21 Subsequently, we investigated the tolerability and the first signs of efficacy of this combination in the phase II dose-expansion cohort.
Anti-angiogenic treatments may influence drug bioavailability. Therefore, we also evaluated the effect of regorafenib administration on paclitaxel pharmacokinetics. 22 To identify the resistance mechanisms and other prognostic or predictive biomarkers, we assessed galectin-1 as a biomarker, previously associated as a biomarker of aberrant tumor vasculature, 23 as well as other potential exploratory biomarker analyses through RNA expression and DNA copy number analyses of metastatic lesions.
Methods
Patient eligibility criteria
Eligible patients were ⩾18 years and had pathologically confirmed metastatic gastric or esophageal adenocarcinomas, refractory to prior cytotoxic treatment including a fluoropyrimidine and platinum compound. Key inclusion criteria were an Eastern Cooperative Oncology Group score of 0/1, adequate bone marrow, renal, and hepatic functions, no suspected brain metastases, and no significant cardiac- or interstitial lung disease (Supplemental Appendix 1.1).
Study design
This was a prospective, multicenter, phase Ib/II dose-escalation study (NCT02406170) with a dose-expansion cohort of regorafenib and paclitaxel in patients with EGC refractory to first- or second-line treatment. Dose escalation was conducted using a standard 3 + 3 design 24 to assess the optimal dose of regorafenib from days 1–21 of a 28-day cycle (dose level 1:80 mg, 2:120 mg, and 3:160 mg), in combination with paclitaxel 80 mg/m2 on days 1, 8, and 15. Regorafenib was administered from days 2–22 in cycle 1 to allow for pharmacokinetic analyses in both cohorts. Dose escalation was permitted if patients were treated with ⩾75% of the assigned dose for 28 days and completed the safety evaluations (dose-escalation rules in Supplemental Table 1). In the dose-expansion cohort, patients received the established MTD of regorafenib on days 1–21.
Dose modifications for toxicity
Toxicity was measured using National Cancer Institute Common Terminology Criteria for Adverse Events version 4.3. Dose-limiting toxicity (DLT) was defined as grade ⩾3 non-hematological toxicity, grade ⩾3 thrombocytopenia, grade ⩾3 infection, or treatment delay of ⩾2 weeks due to treatment-related adverse events. Dose reductions were performed as per the treating physician’s judgment, using the de-escalation rules specified in the protocol (Supplemental Appendices 1.2 and 1.3).
Study endpoints and statistics
The primary objective of this study was to determine the MTD of regorafenib in the dose-escalation cohort and to assess the tolerability of regorafenib with paclitaxel as treatment of patients with EGC refractory to first- or second-line treatment in the dose-expansion cohort. Secondary endpoints were response, OS, and progression-free survival (PFS). Exploratory endpoints were a comparison of OS and PFS with propensity-score matching, paclitaxel pharmacokinetics in relation to safety and survival, and exploratory analyses to identify biomarkers and resistance mechanisms using enzyme-linked immunosorbent assay (ELISA) measurements, RNA expression, and DNA copy number aberrations in relation to response and survival.
As no clinical data are available on the effect of regorafenib on biomarkers and paclitaxel pharmacokinetics, we extrapolated results from pharmacokinetic studies. We assumed an effect size of 0.8 to achieve a power of 80%, which resulted in 26 patients required in the dose-expansion cohort. Accounting for the possibility that a second biopsy could not be performed in 25% of patients, 33 patients were required for inclusion in the dose-expansion cohort. Analyses included all patients who received at least one dose of both study drugs. SPSS Statistics for Windows version 26.0 (IBM Corp., Armonk, NY, USA) was used for statistics. All tests were two-sided, with p < 0.05 considered as statistically significant.
Efficacy
The response was measured according to the RECIST 1.1 criteria. 25 The overall response rate (ORR) was defined as the percentage of patients who demonstrated a complete response (CR) or partial response (PR); disease control rate (DCR) was defined as the percentage of patients who demonstrated CR, PR, or stable disease (SD).
OS and PFS were estimated using Kaplan–Meier analyses, measured from the day of drug administration until documented death of any cause or progression, respectively. Study patients were matched to patients from the Netherlands Cancer Registry (NCR) in a 1:2 ratio using propensity-score matching. Patients were matched on gender, age, performance status, primary tumor location, histological subtype, number of metastases, prior treatment (fluoropyrimidine, platinum, taxane, trastuzumab), time between primary diagnosis and start of palliative treatment or progression, using a logistic regression model. Matching was performed using the nearest neighbor technique. The within-pair difference was minimized by setting a caliper of 0.20 of the standard deviation of the logit of the propensity score. After matching, the balance per item between the groups was assessed by the standardized mean difference. Finally, survival was compared using a Cox proportional hazards model (Supplemental Appendix 1.4).
Paclitaxel pharmacokinetics
For pharmacokinetic analyses, plasma samples were obtained on days 1 and 15 in cycle 1, before paclitaxel administration at t = 0, and 0.5, 1, 1.5, 3, 5, 8, and 24 h after infusion (Supplemental Appendix 1.5). Paclitaxel was extracted from plasma by protein precipitation after the addition of paclitaxel-d5 as an internal standard. Paclitaxel was analyzed using liquid chromatography-mass spectrometry with a detection limit of 1 ng/mL. 26 Concentration versus time data were elaborated according to non-compartmental pharmacokinetic analysis for intravenous infusion. 27 Exploratory analyses were performed on all patients receiving 120 mg regorafenib: dose-expansion cohort (N = 34) and dose-escalation cohort (N = 7). Statistical testing was performed with a paired Wilcoxon test for nonparametric data and paired t-tests for normally distributed data. Correlations with efficacy or safety were analyzed using Spearman’s rho correlation.
Exploratory biomarker analyses
Galectin-1 levels were determined using the Human Galectin-1 Quantikine ELISA kit (R&D systems Europe, Ltd., Abingdon, UK) in duplicate from serum samples obtained on day 1 and day 15. Galectin-1 ratios were calculated by dividing the on-treatment concentration by the baseline concentration. Survival between patients with decreasing (ratio < 1.0) and increasing (ratio > 1.0) levels of galectin-1 were compared using a Cox proportional hazards model.
For RNA expression and genome-wide DNA copy number analysis, fresh-frozen biopsies from metastatic lesions were obtained in the dose-expansion cohort on day 1 and day 15 of cycle 1. RNA and DNA were isolated using the RNeasy Mini Kit (QIAgen AllPrep DNA/RNA/miRNA) following the manufacturer’s protocol. For DNA copy number analyses, fresh-frozen biopsies were unavailable for three patients, and formalin-fixed paraffin-embedded tumor samples were used instead (metastatic biopsies n = 2; primary tumor n = 1) using the DNA FFPE Tissue kit (QIAgen, Hilden, Germany).
RNA expression analyses were performed by RNA sequencing on an Illumina HiSeq 4000, with a single read consisting of 50 bp and 100 million reads per sample. Data were quality controlled using FastQC. Reads were aligned to the human reference genome (NCBI37/hg19) using STAR v2.7.1 and annotated with Gencode v32. The gene set enrichment analyses platform 28 and the R2 genome analysis and visualization platform (http://r2.amc.nl) were used to identify the differential expression of hallmark pathways (MSigDB, 2019) between baseline and on-treatment samples and short (⩽median OS) versus long survivors (>median OS). A gene set of previously established regorafenib targets was used, based on previously published literature. 29 Immune cell deconvolution was performed using the Microenvironment Cell Populations-counter R package. 30 To investigate whether VEGFA was excreted by tumor cells, we performed RNA in situ hybridization VEGFA staining using the RNAscope® Multiplex Fluorescent Kit v2 (Advanced Cell Dagnostics, Newark, CA) according to the supplier's protocol.
Genome-wide DNA copy number analysis was performed by shallow whole-genome sequencing, as previously described. 31 Correction for GC- and mappability bias, dewaving and segmentation were performed accordingly. Calling was performed by comparative genomic hybridization Call, which used the cellularity estimates from an absolute copy number estimator (Supplemental Appendix 1.7).32,33 The association between survival and copy number profiles was analyzed using a two-sided log-rank test with the CGHtest package in R (version 3.4.0). To validate a chromosomal gain or loss, we assessed in R2 if a significant downregulation or upregulation of gene expression on the aberrant chromosomal region of interest was present.
Results
Baseline characteristics
In all, 48 patients were enrolled between July 2015 through November 2018 from three centers in the Netherlands: 14 patients in the dose-escalation cohort and 34 patients in the dose-expansion cohort. The first patient was enrolled on July 27, 2015 (Clinicaltrials.gov, registration number NCT02406170, registered on April 2, 2015). In all, 39 patients were men (81%). The median age of patients was 62 years [interquartile (IQR): 55–69]. The majority of patients had an adenocarcinoma (94%) located in the esophagus (50%), and received one prior line of treatment for metastatic disease (67%; Table 1).
Baseline characteristics.

CT, chemotherapy; CRT, chemoradiotherapy; ECOG, Eastern Cooperative Oncology Group.
Safety and tolerability
In dose level 1 (80 mg/day, n = 3), no DLT occurred. Three patients were enrolled in dose level 2 (120 mg/day), and no DLT was observed. In dose level 3 (160 mg/day, n = 4), three grade 3 DLTs occurred in two patients; diarrhea, gamma-glutamyl transferase increase, and mucositis. Subsequently, four additional patients were enrolled in dose level 2, as one patient was not evaluable for toxicity. In dose level 2, one out of six evaluable patients experienced a DLT: hand–foot syndrome grade 3. Thus, the MTD of regorafenib in combination with paclitaxel was established at 120 mg daily on days 1–21 of each cycle.
In the entire cohort (N = 48), the median number of cycles received was four (IQR: 2–6). The median dose intensity over the first four cycles was 77% for paclitaxel and 87% for regorafenib. Patients discontinued treatment due to disease progression (n = 39), toxicity (n = 7), or at own request (n = 2).
In the entire cohort (N = 48), the most common treatment-related grade 1/2 toxicities were fatigue (75%), peripheral sensory neuropathy (58%), and hoarseness (58%). Of the grade ⩾3 toxicities, hypertension (15%), diarrhea (10%), and pneumonia (10%) were the most prevalent (Table 2). Eight patients (17%) experienced one or more treatment-related serious adverse events (Supplemental Table 2). In two patients, adverse events (pneumonia, suicide due to depression) had a fatal outcome.
Adverse events.

Efficacy
Patients were followed through May 2020; all patients had deceased at this time. With a median follow-up of 7.8 months (IQR: 4.0–10.3), median OS and PFS were 7.8 (95% CI, 6.5–9.2) and 4.2 (95% CI, 3.2–5.1) months, respectively (Figure 1). Six-month OS and PFS were 65% and 27%, respectively.
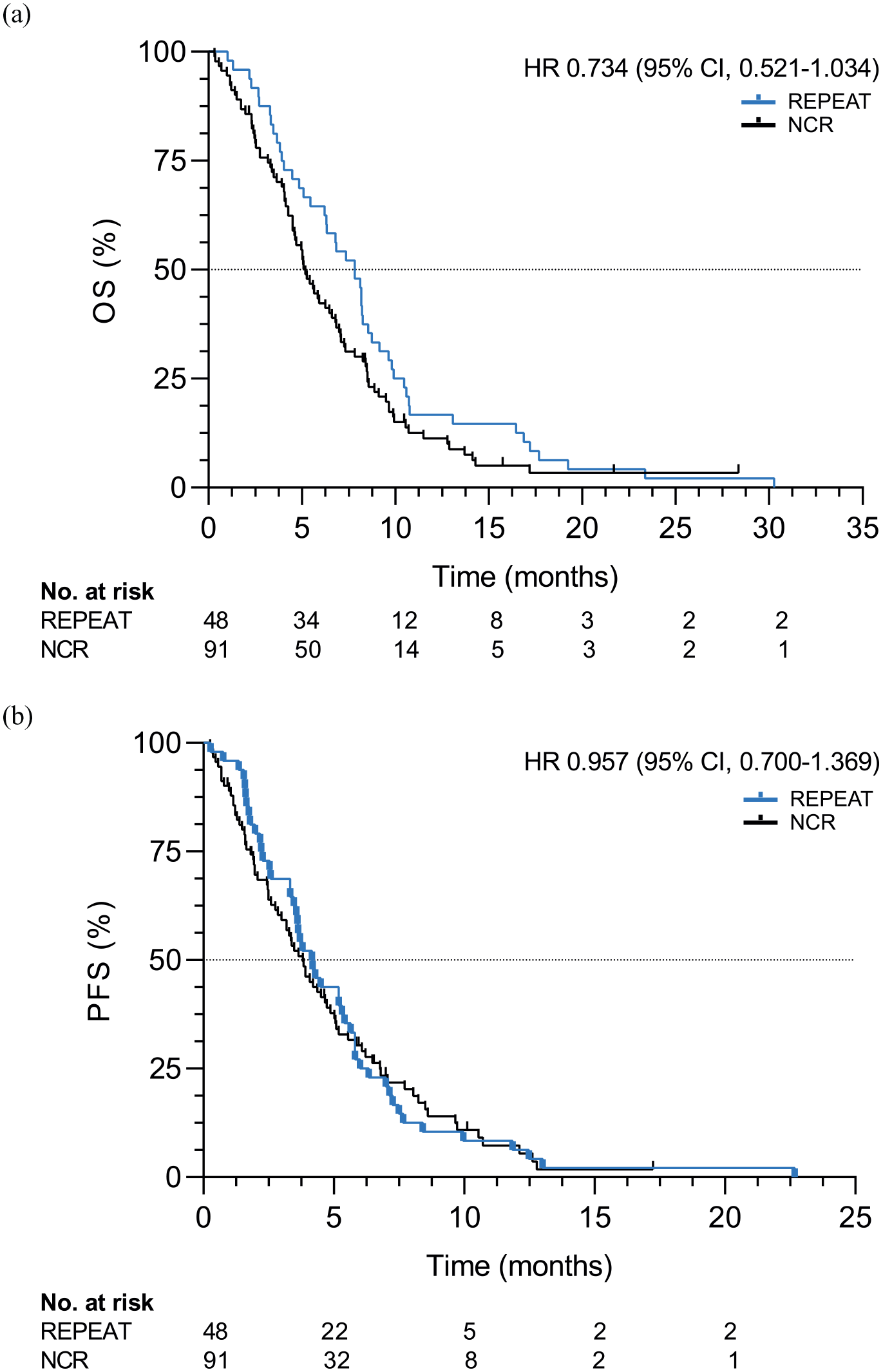
OS (a) and PFS (b) in patients (N = 48) receiving paclitaxel and regorafenib compared to a propensity-score matched cohort of patients (N = 91) from the NCR receiving standard treatment.
All 48 patients were matched to 91 patients from the NCR (Supplemental Table 3). No statistically significant difference in OS was observed in patients receiving paclitaxel and regorafenib compared to the NCR cohort [median OS: 7.8 versus 5.2 months, hazard ratio (HR): 0.73, 95% CI, 0.52–1.03, p = 0.08). Similar results were observed for PFS (median PFS: 4.2 versus 3.8 months, HR: 0.96, 95% CI, 0.67–1.37, p = 0.81; Figure 1).
As two patients died before the first response evaluation scan, 46 patients were evaluated for response. ORR was 28%, and no complete responses occurred. Disease control was achieved in 38 (83%) patients. PR, SD, and PD were achieved in 13 (28%), 25 (54%), and 8 (17%) patients, respectively (Supplemental Figure 1).
Pharmacokinetics
We compared the paclitaxel concentration without regorafenib (day 1) to the concentration with regorafenib (day 15) to assess whether regorafenib influences paclitaxel concentrations in blood. In the dose-escalation cohort, no significant differences in pharmacokinetic parameters of paclitaxel were identified between the cohorts receiving 80, 120, or 160 mg regorafenib (p > 0.05).
Samples were available for analyses of 47 patients. In the entire cohort receiving 120 mg regorafenib (n = 40), a significantly higher maximum concentration (Cmax) of paclitaxel and area under the curve (AUC)0-∞ (median, range) was observed when regorafenib was administered concomitantly compared to paclitaxel only, respectively (Cmax 2865 ng/mL, 367–9610 versus 2485 ng/mL, 499–8360, p = 0.04; AUC0-∞ 5318 ng/mL*h, 1532–11463 versus 4724 ng/mL*h, 1614–14114, p = 0.04; Figure 2, Supplemental Figure 2). Similarly, a significantly longer half-life time of paclitaxel was observed on C1D15 (median: 11.7 h, IQR: 9.6–14.4) compared to C1D1 (median: 9.4 h, IQR: 8.8–11.2; p = 0.001; Supplemental Table 4). The mean time above the threshold paclitaxel concentration (42.7 ng/mL; 0.05 µmol/L) was 11.7 h (SD: 7.10) in C1D1 and 13.7 h (SD: 8.03) in C1D15 (p = 0.306).

Plasma concentrations (ng/mL) of paclitaxel up to 24 h following infusion, measured on baseline without regorafenib co-administration (C1D1) and on-treatment with concomitant regorafenib administration (C1D15) in all patients receiving 120 mg regorafenib (n = 40).
We observed no correlation of pharmacokinetic parameters of paclitaxel with treatment-related toxicity, such as neurotoxicity, mucositis, hand–foot syndrome, or laboratory abnormalities (p values >0.05; Supplemental Table 5). Moreover, no apparent correlation between AUC0-∞ or Cmax was observed with OS (p = 0.916; p = 0.526) or PFS (p = 0.795; p = 0.864).
Exploratory biomarker discovery
Galectin-1 as a biomarker for survival
Galectin-1 has been implicated as a biomarker of treatment resistance to anti-angiogenic therapeutics. 34 Of the 48 patients, 45 patients were included in this analysis based on available serum samples. The median (IQR) galectin-1 serum level from the entire cohort was 39.5 ng/mL (29.3–45.3) on day 1 and 40.0 ng/mL (9.8–53.1) on day 15. In our study, patients with increased galectin-1 levels on-treatment compared to baseline [ratio > 1.0; 46.4 ng/mL (38.8–54.9) day 15 versus 31.0 ng/mL (26.8–44.1) on day 1] had a shorter OS compared to those with decreased levels [31.2 ng/mL (24.6–40.1) versus 41.3 ng/mL (39.2–56.2); HR 0.49 (95% CI, 0.27–0.90), p = 0.008, Figure 3(a)].

(a) OS in patients with an increase in galectin-1 levels on day 15 compared to day 1 (ratio >1.0; n = 24) versus a decrease (ratio < 1.0; n = 21) measured with ELISA. (b) A heatmap of a gene panel from previously established regorafenib targets on baseline (day 1, n = 21) compared to on-treatment (day 15, n = 13) using RNA sequencing. (c) Normalized enrichment score of established hallmark pathways (MSigDB, 2019) on-treatment (day 15, n = 13) compared to baseline (day 1, n = 21) according to Gene Set Enrichment Analysis in patients with a short survival and long survival. (d) OS in patients with a gain (n = 12) of chromosome 19q13.12-q13.2 versus no gain (n = 22). (e) Validation of chromosome 19q13.12-q13.2 in RNA sequencing dataset in patients with a gain (n = 12) versus no gain (n = 22) compared to a random region on chromosome 19p13.2-13.13.
Gene expression
As regorafenib has a complex inhibition pattern, we investigated the expression of previously established regorafenib targets and the hallmark pathway angiogenesis using RNA sequencing on metastatic tumor biopsies from 23 patients (21 baseline and 13 on-treatment biopsies). Overall, no significant difference in the expression of reported regorafenib targets or hallmark pathways was observed after 15 days of treatment (Figure 3(b), p = 0.172). A significant increase in the expression of CD8+ cells, B cells, natural killer cells, and neutrophils was observed in the on-treatment samples compared to baseline (Supplemental Figure 4). However, no correlation between survival or response and the immune deconvolution data was observed (Supplemental Figure 4; Supplemental Table 6). Subsequently, we compared patients with short OS to patients with long OS. In patients with short survival, we observed an increased enrichment of several pathways on-treatment compared to baseline, of which MTORC1, spermatogenesis, Wnt, UV response, and angiogenesis showed the strongest increase (Figure 3(c)). From the genes involved in these pathways, only VEGFA expression was significantly associated with OS and PFS. Patients with high baseline expression of VEGFA demonstrated a shorter OS [HR: 0.27 (95% CI, 0.10–0.72), p < 0.001] and PFS [HR 0.40 (95% CI, 0.17–0.96)] compared to those with low baseline VEGFA expression (p = 0.006; Supplemental Figure 3). RNA in situ hybridization analyses confirmed that VEGFA was expressed predominantly in tumor cells rather than endothelium or immune cells (Supplemental Figure 6).
DNA copy number analysis
Copy number variations have previously demonstrated predictive value in a large cohort of metastatic colorectal cancer patients treated with the anti-angiogenic agent bevacizumab. 35 Using genome-wide copy number profiles performed for 34 patients, we identified four prognostic chromosomal regions in chromosomal bands 9q21.1-q31.2, 12q12, 19q13.12-q13.2, and 21q22.11-q22.3 (exact chromosomal locations in Supplemental Appendix 1.7; Supplemental Figure 5). We analyzed if gene expression levels were associated with the specific lost or gained chromosomal regions. This only held for chromosome 19, with significantly higher expression of genes expressed on region 19q13.12-q13.2 in patients with a gain compared to patients without a gain (p = 0.032, Figure 3(d)–(e)). No significantly higher expression was observed between patients with a gain versus no gain when selecting another random region on chromosome 19 (p = 0.874, Supplemental Figure 5). In patients with amplification (24% 2+; 12% 1+) of chromosome 19q13.12-q13.2, a shorter OS [HR: 0.444 (95% CI, 0.190–1.037), p = 0.015] and PFS [HR: 0.467 (95% CI, 0.202–1.077), p = 0.024] were observed compared to patients without amplification (59% 0, 6% −2). Galectin-4, a promotor of angiogenesis associated with gastric cancer progression, is expressed on 19q13.12-q13.2. 36 In the RNA sequencing data, galectin-4 demonstrated a correlation to poor outcome, with higher levels in patients with a shorter OS (p = 0.007) and PFS (p = 0.019). Thus, these results confirm that chromosomal gains could play a role in sensitivity mechanisms.
Discussion
This is the first study to investigate the tolerability of paclitaxel and regorafenib in patients with advanced EGC refractory to first-line treatment. The combination of paclitaxel and regorafenib was shown to be well tolerated, with an established MTD of 120 mg regorafenib per day.
We observed a similar toxicity profile compared to studies administering regorafenib in several other solid tumors, with manageable adverse events. Similar or lower incidences of gastrointestinal and regorafenib-related toxicities, such as hand–foot syndrome, were observed in our study.9 –14,18,19,37,38 Moreover, less hematologic toxicity was seen in our study compared to studies with regorafenib monotherapy.10,18,37 In contrast, higher incidences of peripheral neuropathy and alopecia were observed compared to the aforementioned trials, reasonably attributable to the combination with paclitaxel. Adverse events associated with anti-angiogenic treatment, such as hypertension, epistaxis, and gastrointestinal hemorrhage, were comparable to a large study administering paclitaxel and the VEGFR-2 antagonist ramucirumab. 17 Although it has been reported that the combination of targeted therapy with cytotoxic chemotherapy increases toxicity and discontinuation rates, the current treatment combination did not seem to result in more toxicity than that was reported with both drugs individually.19,20
With a median OS of 7.8 months compared to 5.2 months in a propensity-score matched real-world cohort, we observed a numerically higher OS, albeit not statistically significant. Although OS in our study was higher than the majority of second- or third-line studies in EGC,4,15,39 –41 a longer median OS (9.6 months) was observed in patients with gastric adenocarcinoma treated with ramucirumab as second line in the RAINBOW study. 17 Nevertheless, it should be noted that one-third of patients included in our study received treatment as third line. DCR was similar to the RAINBOW study, and treatment with paclitaxel and regorafenib resulted in higher ORR and disease control than other second- or third-line studies.4,15,17,41
Significantly higher concentrations of paclitaxel were observed when administered concomitantly with regorafenib. Overall, both paclitaxel concentrations and time above the minimum threshold were comparable to the literature.42,43 Pharmacokinetic parameters of paclitaxel have been associated with increased incidences of adverse events. This was not observed in our study, which could support the tolerability of the paclitaxel dose with regorafenib co-administration. 43 Similar to our results, previous research also demonstrated a prolonged duration of plasma chemotherapeutic exposure of docetaxel with concomitant administration of bevacizumab in lung cancer patients. 44 However, in this study, higher peripheral chemotherapeutic concentrations did not result in increased intratumoral chemotherapeutic uptake. As literature is still indecisive whether anti-angiogenic treatment increases intratumoral drug delivery through normalization of the aberrant tumor vasculature, the impact of regorafenib on intratumoral drug delivery remains to be established.
Since we only observed a trend toward a beneficial effect in our unselected cohort, it is essential to identify the mechanisms of resistance and predictive biomarkers to better tailor treatment to individual patients. Due to the intricate inhibition pattern of regorafenib, the identification of predictive biomarkers is challenging. 45 No direct effect of regorafenib administration was observed on previously established regorafenib targets in the gene expression data. The lack of observed effect may be explained by the short time interval between the first and second biopsies, although both intratumor and cohort heterogeneity could also play a role in this relatively small cohort. However, we did observe increased enrichment during treatment predominantly in patients with a short survival compared to those with a long survival. This suggests that alternative activation of angiogenesis in the presence of regorafenib could account for the difference in response. Interestingly, it has been shown that galectin-1 can counteract VEGF-targeted therapy by maintaining VEGFR2 signaling even when VEGF is blocked. 46 Moreover, previous literature has demonstrated that galectin-1 is a potent angiostimulatory protein, and circulating galectins can serve as biomarkers of therapeutic efficacy.23,47 In our cohort, patients with increasing galectin-1 upon treatment with regorafenib and paclitaxel show a worse outcome when compared to those with decreasing levels, which may reflect compensatory pro-angiogenic signaling. These data coincide with the observation that patients with a high VEGFA tumor expression at baseline, an important ligand of VEGFR1 and VEGRF2, had poorer outcomes compared to lower VEGFA levels, as also published previously.48,49
In our study, a gain of chromosome 19q13.12-q13.2 demonstrated a prognostic value. The potential value as biomarker of this chromosomal region (19q) has previously been identified in esophageal tumors. 50 These findings could indicate the presence of a target gene involved in EGC tumorigenesis on chromosome 19q13.12-q13.2. It is thought that galectin-4, expressed on the latter region and previously associated with galectin-1, plays a role in anti-angiogenic mechanisms by contributing to tumor angiogenesis through enhancement of endothelial cell activity. 51 As both galectin-1 and galectin-4 have been correlated with short survival in patients treated with paclitaxel and regorafenib, these findings could potentially confirm the role of galectins in resistance to regorafenib. However, further investigation is necessary to identify whether it could serve as a predictive biomarker for treatment with regorafenib.
In addition to the presumed role of galectins in anti-angiogenic mechanisms, previous research also unraveled immunoregulatory properties of the galectin family. 52 Galectin-1 and galectin-4 are assumed to play a role in mediation of the suppressive function of regulatory T cells. 52 Unfortunately, we did not observe a statistically significant correlation between response or survival and immune cell expression. Further research could demonstrate whether patients with high expression of galectins, and thus potentially less response to regorafenib, could benefit from the combination of anti-angiogenic treatment with regorafenib and immunotherapy to normalize vascular-immune crosstalk. 53
In conclusion, treatment with paclitaxel and regorafenib was tolerable and safe in patients with advanced EGC refractory to first-line systemic therapy. Efficacy results were promising, and a numerical, non-significant survival benefit was also demonstrated compared to a propensity-score matched cohort. Galectin-1 and a gain of chromosome 19q13.12-q13.2 are promising negative prognostic, and potentially predictive biomarkers for paclitaxel and regorafenib. These biomarkers could play a role in selecting patients with most benefit for treatment with regorafenib and paclitaxel in future studies. Further research in a randomized controlled trial is warranted to validate these findings.
Supplemental Material
sj-pdf-1-tam-10.1177_17588359221109196 – Supplemental material for A phase Ib/II study of regorafenib and paclitaxel in patients with beyond first-line advanced esophagogastric carcinoma (REPEAT)
Supplemental material, sj-pdf-1-tam-10.1177_17588359221109196 for A phase Ib/II study of regorafenib and paclitaxel in patients with beyond first-line advanced esophagogastric carcinoma (REPEAT) by Charlotte I Stroes, Sandor Schokker, Mohammed Khurshed, Stephanie O van der Woude, Ron AA Mathôt, Marije Slingerland, Judith de Vos-Geelen, Massimo Zucchetti, Cristina Matteo, Erik van Dijk, Bauke Ylstra, Victor Thijssen, Sarah Derks, Tesfay Godefa, Willemieke Dijksterhuis, Gerben E Breimer, Otto M van Delden, Rob HA Verhoeven, Sybren L Meijer, Maarten F Bijlsma and Hanneke WM van Laarhoven in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
We would like to acknowledge all patients who participated in this study, as well as their families. We would also like to thank all nursing staff from the Amsterdam UMC, and M.J. Weterman for her essential support. Furthermore, we would like to thank M. Steketee for his support in the DNA copy number analyses, as well as H.F.B. van Essen for the NGS copy number profiling laboratory experiments.
Ethics approval and consent to participate
This study was approved by the institutional review board of the AmsterdamUMC (METC, 2015_013) and conducted following the guidelines of Good Clinical Practice and the ethical principles of the Declaration of Helsinki. All patients provided written informed consent.
Consent for publication
Not applicable.
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this investigator-initiated study was derived from the Amsterdam UMC, location AMC, with financial support through an unrestricted research grant from Bayer BV.
Conflict of interest statement
All authors completed the disclosure of conflict of interest. The following authors declared a potential conflict of interest: S. Schokker has received travel and accommodation reimbursement from Roche. R.A.A. Mathôt has received grants from NOW, ZonMW, and unrestricted investigator research grants from Baxter, Baxalta, Shire, Takeda, Bayer, CSL Behring and Sobi, and reports and advisory role for Bayer, CSL Behring, Merck Sharp & Dohme, Baxter, Baxalta, Shire and Takeda, with fees paid to the institution. R.H.A. Verhoeven has received research grants from Roche and BMS. M.F. Bijlsma reports an advisory role for Servier, and has received a research grant from Celgene. M.I. van Berge Henegouwen reports unrestricted research grants from Stryker and Olympus with fees paid to the institution, and an advisory role for Johnson and Johnson, Mylan, and Medtronic, with fees paid to the institution. H.W.M. van Laarhoven reports an advisory role for BMS, Celgene, Lilly, Merck, Nordic, and Servier, and has received unrestricted research funding from Bayer, BMS, Celgene, Lilly, Merck Serono, MSD, Nordic, Philips, Roche and Servier, with fees paid to the institution.
Supplemental material
Supplemental material for this article is available online.
Data availability statement
The sequence data generated in this study are made publicly available in the following archives, and are available within the article and its supplementary data files:
- The shallow whole-genome sequencing data have been deposited at the European Genome-phenome Archive (EGA), under accession number EGAS00001006054.
- The RNA sequencing data has been deposited at the Gene Expression Omnibus database (GEO), under accession number GSE198136.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.