
Abstract
Background:
Advances in targeted therapy development and tumor sequencing technology are reclassifying cancers into smaller biomarker-defined diseases. Randomized controlled trials (RCTs) are often impractical in rare diseases, leading to calls for single-arm studies to be sufficient to inform clinical practice based on a strong biological rationale. However, without RCTs, favorable outcomes are often attributed to therapy but may be due to a more indolent disease course or other biases. When the clinical benefit of targeted therapy in a common cancer is established in RCTs, this benefit may extend to rarer cancers sharing the same biomarker. However, careful consideration of the appropriateness of extending the existing trial evidence beyond specific cancer types is required. A framework for extrapolating evidence for biomarker-targeted therapies to rare cancers is needed to support transparent decision-making.
Objectives:
To construct a framework outlining the breadth of criteria essential for extrapolating evidence for a biomarker-targeted therapy generated from RCTs in common cancers to different rare cancers sharing the same biomarker.
Design:
A series of questions articulating essential criteria for extrapolation.
Methods:
The framework was developed from the core topics for extrapolation identified from a previous scoping review of methodological guidance. Principles for extrapolation outlined in guidance documents from the European Medicines Agency, the US Food and Drug Administration, and Australia’s Medical Services Advisory Committee were incorporated.
Results:
We propose a framework for assessing key assumptions of similarity of the disease and treatment outcomes between the common and rare cancer for five essential components: prognosis of the biomarker-defined cancer, biomarker test analytical validity, biomarker actionability, treatment efficacy, and safety. Knowledge gaps identified can be used to prioritize future studies.
Conclusion:
This framework will allow systematic assessment, standardize regulatory, reimbursement and clinical decision-making, and facilitate transparent discussions between key stakeholders in drug assessment for rare biomarker-defined cancers.
Keywords
Introduction
Advances in high-throughput sequencing technology and improved understanding of molecular drivers of carcinogenesis 1 continue to identify potentially targetable molecular alterations and pathways.2–4 As a result, the drug development paradigm is shifting to include approaches that match targeted therapies to specific molecular alterations. Effectiveness of targeted therapy in one histology in the presence of a matching molecular alteration, also referred to herein as a “predictive biomarker,” might then establish a rationale for classifying other cancer types with the same biomarker, as a potentially histology-independent, druggable target (Box). In this way, cancers may be classified into smaller subgroups.
Glossary of terms.

Small population size presents a critical practical challenge for generating robust evidence within an acceptable timeframe for regulatory approval and reimbursement of novel targeted therapies. Typically, novel therapies are assessed through comparison with standard-of-care treatment on measures of net clinical benefit, such as overall survival (OS) and quality of life, from randomized controlled trials (RCTs).5,6 However, adequately powering RCTs for these measures for each biomarker-defined cancer subgroup may be infeasible. Thus, there are calls for approval of molecularly targeted therapies to be based on non-randomized studies, including basket studies, using intermediate endpoints such as objective response rates (ORRs).7–9 Tempering these calls, reviews of phase III RCTs have shown the clinical benefit of molecularly targeted therapies is often only modest when compared with standard-of-care non-targeted therapies.10–12 Further, post-approval commitments and timely withdrawal of drugs do not always occur when required, potentially exposing patients to therapies that are less effective and/or more harmful than previously assumed.13–15 Uncontrolled histology-agnostic studies provide valuable proof-of-concept evidence for rare cancer populations. However, without randomized comparison, it is difficult to differentiate the clinical benefit of targeted treatment from the natural course of the disease process (“natural history”) for each cancer type, other biases such as selection bias and especially to distinguish any predictive properties from any prognostic properties of the biomarker for each cancer type (Box). RCTs in rare cancers, albeit on intermediate or surrogate outcomes, are ideal and recommended wherever possible. Innovative trial designs are being developed to increase study power to assess the predictive value of a biomarker and include biomarker-adaptive designs using classical or Bayesian methods for randomization according to biomarker status and treatment group.16–19 However, as demonstrated by regulatory decisions based on non-randomized evidence, 20 there is an immediate need for a framework to support transparent decision-making where robust RCT data on clinically relevant outcomes is not available.
In this paper, we address the problem where RCT evidence of the effectiveness of targeted treatment is available for at least one cancer type (referred to herein as the “common cancer”), and the question is whether it can also be recommended in other cancers sharing the same biomarker for which a new RCT is not feasible on clinically relevant outcomes due to small population size (referred to herein as the “rare cancer”). In this context, extrapolation refers to the leveraging or extending process whereby an indication for use of a therapy in a new patient population can be supported by existing clinical data from a related studied patient population.21–23 Extrapolation of evidence from drugs already approved in adults is accepted to support submissions for pediatric use provided that the disease course and treatment response are sufficiently similar in both populations.22,23 Similarly, in rare biomarker-defined cancer populations, it may be possible to extrapolate evidence from similar populations where robust RCT evidence exists for the same targeted therapy to support regulatory approval in the rare cancer. 21 However, careful consideration of the appropriateness of extending the existing trial evidence beyond specific cancer types is required.
In this framework, we outline a series of questions to guide extrapolation of evidence for a molecularly targeted therapy generated from RCTs in common cancers to different rare cancers sharing the same biomarker.
Extrapolation framework
Current approach for evaluation
RCTs enrolling patients with biomarker-positive and biomarker-negative disease to compare the targeted therapy against standard non-targeted care can provide definitive evidence to assess treatment effectiveness in both groups and distinguish the prognostic and predictive value of a biomarker (Figure 1(a)). For targeted therapies in the common cancer, RCTs may provide this comprehensive evidence. Where data is restricted to the biomarker-positive population, stronger evidence for treatment efficacy and/or evidence across a range of cancer types is required for extrapolation. For rare biomarker-defined cancers, targeted therapies are typically assessed in single-arm studies in the biomarker-positive population (Figure 1(b)). In this scenario, pragmatic cross-study comparisons with a historical control group are usually relied upon to support claims of treatment effectiveness. Ideally, prognostic studies in patients with the rare cancer, treated with the same standard (non-targeted) treatment or best supportive care, and known biomarker status would be available for these comparisons (Figure 1(c)). These studies allow assessment of both the prognostic value of the biomarker in the rare cancer by comparing outcomes for biomarker-positive and biomarker-negative patients; and provide an untreated historical control group for cross-study comparison with the single-arm study/studies of the targeted therapy to determine treatment effectiveness. Our previous scoping review of methodological guidance showed regulatory agencies, health technology assessment (HTA) bodies, research groups, and others use different approaches for this assessment. 24 Each group incorporated additional topics to guide extrapolation of evidence from common to rare cancers, but we did not identify a framework to promote the explicit assessment of commonly used criteria.

Approach for evaluating biomarker-targeted therapies in common and rare cancers.
Extrapolation framework
We developed the framework from the core topics for extrapolation identified from the scoping review 24 and incorporated the principles outlined in the medicines extrapolation framework of the European Medicines Agency. 21 The framework is presented as a series of questions articulating essential criteria for extrapolation, with illustrative examples. The criteria reflect key assumptions of similarity for the disease definition and treatment outcome between the common and rare cancer (Table 1). To support the assumption of similar treatment outcomes, extrapolation can be more readily considered if the targeted treatment is proposed as last-line therapy in the rare cancer. Extrapolation would be more complex if effective alternative therapies existed. We have further outlined a pragmatic approach for evaluating existing evidence to judge these criteria for regulatory approval, reimbursement, and clinical decisions (Figure 2). The level of uncertainty for each criterion can be judged based on existing approaches for evidence-based decision-making. 25 Each criterion should be considered individually and then the overall assessment should be made based on the totality of the evidence to judge whether adequate to conclude treatment effectiveness and favorable benefit–risk profile 26 in the rare cancer. The evidence available for each criterion may increase or decrease uncertainty for the overall judgment (Table 2). Knowledge gaps where limited evidence exists should inform future research to acquire additional data. The framework is intended to describe the criteria that need to be explicitly addressed for decision-making. It is not intended to be prescriptive since the suitability for data extrapolation will likely vary for different biomarker-targeted therapy-cancer scenarios. The reporting of this study conforms to the RIGHT statement modified for a research framework 27 (Supplemental Material).
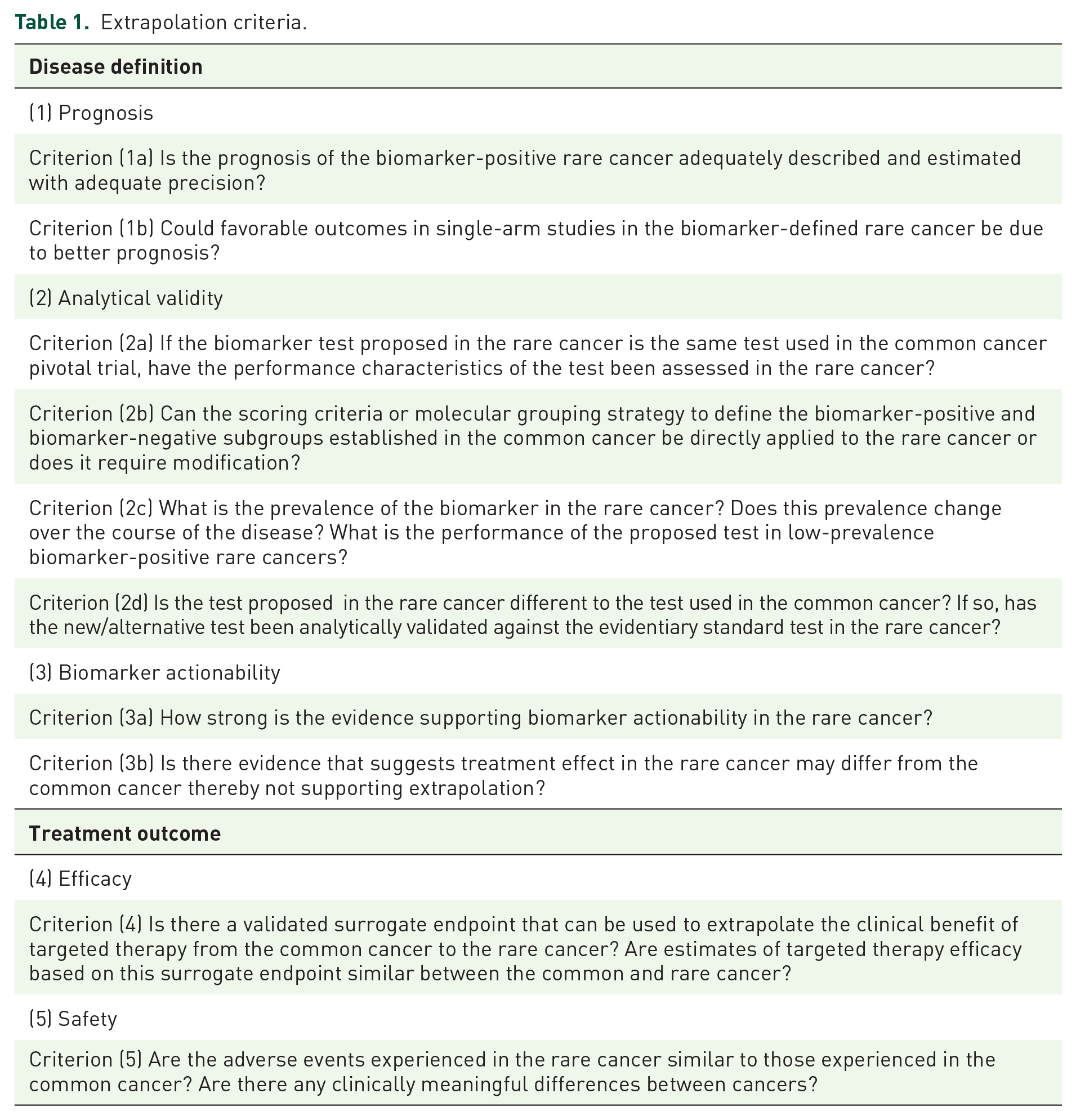
Extrapolation criteria.

Extrapolation framework: decision tree.
Assessment of uncertainty when extrapolating evidence for transparent decision-making.

Source: Adapted from Piggott et al. 28
Judgment for the level of uncertainty for extrapolation should be made individually for each criterion. Judgments from other extrapolation criterion either increase or decrease certainty of each criterion.
The final decision should be made based on the totality of the evidence. If there is probably no important uncertainty for most of the criteria, then there is likely sufficient evidence to support regulatory approval. Any substantial knowledge gaps identified resulting in possibly important uncertainty for one or more criteria should define additional studies required pre-approval. If there is important uncertainty for many or most of the criteria, further studies are required to address knowledge gaps for later reassessment. Judgment for decision-making should be individualized and consider estimated benefits versus risks of targeted therapy compared to alternative therapies if available.
Components
We propose assessing disease similarity under three extrapolation components: (1) “Prognosis” addressing clinical outcomes of biomarker-defined cancer in the absence of targeted treatment to inform control data, (2) “Analytical Validity” addressing the performance characteristics of the test used to identify the biomarker, and (3) “Biomarker Actionability” addressing the evidence that the biomarker represents a dominant targetable molecular pathway and predicts the effect of the therapy being assessed. We propose assessing similarity of treatment outcomes under two components: (4) “Efficacy” addressing predictions of similar clinical benefit between cancers based on signals of efficacy on intermediate/surrogate outcomes in the rare cancer and (5) “Safety” addressing similarity of the safety profile between cancers and methods to augment safety data in the rare cancer. The order of the components is not fixed, and a pragmatic approach may be to start with identifying the best available evidence of treatment outcomes in the rare cancer first and addressing other criteria later.
Disease definition
Prognosis
Criterion (1a) Is the prognosis of the biomarker-positive rare cancer adequately described and estimated with adequate precision for use as a historical control?
The prognosis of biomarker-positive rare cancers describes the natural history in the absence of targeted therapies. Natural history may be available for the histology-defined cancer without biomarker information. However, targeted therapies are developed to reverse or inactivate an aberrant biological pathway and the related biomarker may be associated with unfavorable, favorable, or neutral prognosis. For example, HER2 gene amplification or overexpression is a poor prognostic factor in breast cancer 29 and anti-HER2 therapies, such as trastuzumab, have been shown to reverse the natural history of this poor prognosis disease. 30
Historical control data for the biomarker-positive population could be obtained from retrospective biomarker analyses of RCTs or cohort studies testing non-targeted therapy, and real-world studies annotated with biomarker data (e.g. electronic health record data or registries). Critical requirements for such prognostic studies include unbiased patient selection, large sample size, uniform treatment, high-quality data collection for marker status at baseline, identification of potential confounders, complete and long-term follow-up for clinical outcome assessment, and outcome ascertainment with sufficient precision and replicability.31,32 The Reporting Recommendations for Tumor Marker Prognostic Studies checklist for reporting prognostic marker studies details important issues for study design and conduct.33,34
When natural history data are only available for the rare cancer type without biomarker stratification, extrapolating data on prognosis of the biomarker-positive tumor from the common to rare cancer might provide the best available evidence but it is associated with high level of uncertainty. Statistical modeling techniques such as propensity score matching to generate synthetic control arms, and adjusting for known prognostic factors including differences in histotypes, could be used to better estimate prognosis, but such approaches are still limited as it is not possible to account for all possible confounders.35–43
Criterion (1b) Could favorable outcomes in single-arm studies in the biomarker-defined rare cancer be due to better prognosis?
Evidence that a biomarker has no or worse prognostic impact in rare cancer provides greater confidence that favorable outcomes from a single-arm study may be attributable to the targeted therapy. Even so, other biases, such as selection bias, may lead to better outcomes in single-arm studies. Any given biomarker may be prognostic but not predictive, predictive but not prognostic, both prognostic and predictive, or neither prognostic nor predictive. HER2 overexpression in breast cancer is an example of a biomarker that is both prognostic and predictive.29,30 When biomarker expression is associated with good prognosis, such as in the case of hormone receptor-positive breast cancer, the benefit of targeted treatments will be difficult to establish in the absence of RCTs. Favorable clinical outcomes from single-arm studies are often assumed to be the effect of the targeted therapy but may, in fact, be due to the indolent natural history of cancer.
Analytical validity
Analytical validity refers to the analytical performance characteristics of a test to reliably detect the biomarker in a biological specimen. Measures include concordance, sensitivity, and specificity against a validated test, and reproducibility. Assessment of the analytical validity of the biomarker test (assay/technology) is distinct to, but predicated on, the clinical utility of the biomarker to predict treatment benefit. The pivotal RCTs that established targeted treatment effectiveness in the common cancer, also establish the clinical utility of the biomarker in the common cancer. 44 As such, the biomarker test used in the pivotal RCT is generally regarded as the “clinical utility standard” (or evidentiary standard) test for assessment of analytical performance. However, the analytical performance characteristics of the test established in common cancers may or may not be directly relevant to rare cancers.44–46 Two central issues are: first, without an RCT to validate the biomarker predicts treatment benefit in the rare cancer, assessing the analytical validity of the biomarker test for use in the rare cancer will be more complex. Second, as technology evolves, the biomarker test proposed in the rare cancer may not be the same as that used in the pivotal RCT in the common cancer. These and other issues that should be considered when evaluating analytical validity of the biomarker test are outlined below:
Criterion (2a) If the biomarker test proposed in the rare cancer is the same test used in the common cancer pivotal trial, have the performance characteristics of the test been assessed in the rare cancer?
The test proposed in the rare cancer may be the same test used in the RCT of the common cancer. Pre-analytic factors that affect quality of analytes include specimen type (e.g. core tumor biopsy vs blood), preservation (e.g. fresh vs formalin-fixed paraffin-embedded), tissue fixation methods (e.g. time to fixation, duration and temperature of fixation, fixing agent), and specimen age. These factors are specified for the clinical utility standard test and influence the usefulness of the assay. Even so, biological differences between the cancers may alter the test’s performance characteristics, potentially limiting applicability in rare cancers. For example, excessive melanin pigment in some melanomas can interfere with DNA polymerases used in polymerase chain reaction (PCR) methods and invalidate test results. 47 Testing should be undertaken in accredited laboratories. Sufficient concordance and reproducibility across laboratories should be confirmed.
Criterion (2b) Can the scoring criteria or grouping strategy to define the biomarker-positive and biomarker-negative subgroups established in the common cancer be directly applied to the rare cancer or does it require modification?
Scoring criteria
For some binary biomarkers such as DNA point mutations, the same criteria to define the biomarker-positive and biomarker-negative subpopulations in one cancer could be directly applied to another. For example, in the Kirsten rat sarcoma viral oncogene homolog gene, a single-nucleotide variation, where glycine is substituted by cysteine at codon 12 (KRAS G12C), results in activation of downstream signaling pathways. This mutation is found in some non-small cell lung cancers (NSCLC),48,49 colorectal cancers (CRC), 50 and pancreatic adenocarcinoma 51 and the same criteria could be used to classify patients across the different cancer types.
For other biomarkers, such as some quantitative biomarkers or gene signatures, existing criteria in one cancer type will always need to be modified for use in another cancer type. For example, in breast cancer, HER2 gene amplification induces HER2 protein overexpression on tumor cell membrane and is known to be oncogenic.52,53 Although no “gold” standard exists for detecting HER2 alterations, 54 the scoring algorithm based on HER2 amplification, using HER2 gene copies per nucleus or the HER2 gene signals to chromosome 17 centromere ratio as detected by fluorescent or silver in situ hybridization, and HER2 protein overexpression as detected on immunohistochemistry, have been widely validated in breast cancer as a predictive biomarker for various HER2-targeted therapies.54–56 This scoring algorithm required modification before applying to gastric/gastroesophageal junction cancers due to differences in pattern of HER2 expression.57,58 The HER2 scoring systems for CRC 59 and endometrial serous carcinoma 60 are also modified and differ slightly from each of the other cancers.
Grouping strategy
Different but related molecular alterations involving one or more genes affecting a common pathway can result in the same clinical disease. 61 The grouping of alterations may be accepted if there is strong rationale that the group will respond similarly to therapy based on clinical, preclinical, or in silico (computational) mechanistic evidence. 61 In this way, where various molecular alterations comprise the biomarker-defined disease in the common cancer, the same grouping strategy may be used to define the disease in the rare cancer. To illustrate, numerous “deleterious” mutations of the breast cancer susceptibility genes 1 and 2 affect a common DNA repair pathway resulting in a similar phenotype that predicts treatment benefit with poly(adenosine diphosphate–ribose) polymerase enzyme inhibitors in breast, ovarian, prostate, and pancreatic cancers.62–66 Depending on the strength of scientific rationale, it may be reasonable to either expand or restrict the alterations included in the common cancer trial when applying the data to the rare cancer.
Establishing databases of rare cancers annotated with comprehensive genomic profile data would be very useful for assay development and validation of scoring. The criteria or grouping strategy to define the biomarker-positive and biomarker-negative subgroups in the rare cancer should initially be established a priori by consensus based on available information from common cancers. Rare cancer databases that also capture the natural history and clinical outcomes of targeted therapies can also be used to validate the biomarker criteria established in the rare cancer. Modification of criteria may be necessary depending on findings from validation studies (4).
Criterion (2c) What is the prevalence of the biomarker in the rare cancer? Does this prevalence change over the course of the disease? What is the performance of the proposed test in low-prevalence biomarker-positive rare cancers?
Biomarker prevalence can vary widely across different cancer types, stages, treatments, and disease trajectories. 67 For example, mismatch repair deficiency (dMMR) results from mutations in a family of genes involved in DNA repair. This biomarker is considered to be predictive of immunotherapy benefit, and pembrolizumab, a programmed death 1 (PD-1) inhibitor, has been approved for solid tumors with dMMR following progression on prior treatment.68,69 The prevalence of dMMR varies widely across histotypes, ranging from approximately 28% in endometrial cancer 70 to 0.04% in breast cancer. 71 Furthermore, within the same cancer, such as CRC, prevalence of dMMR can also vary between early-stage disease (10%–20%) and advanced-stage disease (3%–4%). 72 Biomarker status can also change over the course of the disease as part of the disease trajectory and/or result of previous treatment.44,73
For a test with a given analytical sensitivity and specificity, changes in biomarker prevalence can significantly alter its positive predictive value (PPV) and negative predictive value (NPV) 44 (Box). A test with high sensitivity and specificity will have poorer PPV in cancers where biomarker prevalence is low compared to other cancers with higher prevalence of the same biomarker. The same test will have poorer NPV in cancers where biomarker prevalence is high compared to cancers with lower biomarker prevalence.45,73 Incorrect classification of a patient (a false positive or false negative result) can potentially result in incorrect treatment recommendations. 73
The prevalence range of the biomarker in the rare cancer as determined by the proposed test for the disease setting should be assessed. The PPV and NPV of the test can be calculated using estimates of sensitivity and specificity. 44 In rare cancers with low biomarker prevalence, a test with sensitivity and specificity approaching 100% should be used whenever possible to minimize the false negative and false positive rates, respectively. 73 Tolerance of a higher false positive rate would depend on the potential for treatment harm, treatment costs, and delays to more effective alternative therapies if available.
Criterion (2d) Is the test proposed in the rare cancer different to the test used in the common cancer? If so, has the new/alternative test been analytically validated against the evidentiary standard test in the rare cancer?
With advancements in diagnostic technology following the pivotal treatment trial in the common cancer, a new test may be considered a more valid measure of the biological target. The new test proposed to identify the biomarker in the rare cancer may use similar (e.g. two different commercially developed PCR tests) or different (e.g. panel point mutations vs whole exome sequencing) technology. When the test proposed for the rare cancer is not the same test used in the common cancer, it may result in discordance between the biomarker-defined populations using each test. 74 Retrospective testing of patient samples from the pivotal trial to assess concordance with an accepted clinical utility standard test and linked with clinical outcome data is ideal and should be done if possible75,76 but may not be feasible. 77 Concordance measures include positive percent agreement, negative percent agreement, and overall percent agreement (Box). However, different organizations have adopted different criterion and the extent of sufficient agreement is an unresolved issue.78–80 Discrepancies are resolved using another orthogonal method. 77 Intra-observer, inter-observer, and inter-laboratory reproducibility is assessed where appropriate.45,81 Where discordance exists between the two tests, there would be insufficient evidence of clinical utility of the biomarker as defined by the new test in rare cancer.
Biomarker actionability
A biomarker is potentially “actionable” if it represents (i) a molecular pathway driving oncogenesis and tumor progression that can be mitigated or reversed by targeted therapy to improve clinical outcomes and is not also affected by (ii) a resistance pathway so that targeted therapy rapidly becomes ineffective. Critical to demonstrating actionability is evidence of the ability of the biomarker to predict clinical outcomes. Methods to validate a predictive biomarker within a specific cancer type utilizing trial designs to assess for the biomarker-treatment interaction are established. 82 When assessing the predictive value of a biomarker in rare cancers utilizing Bayesian adaptive trial designs, increased biological understanding may reasonably shift Bayesian priors. In this paper, we assume the predictive value of the biomarker has been validated in the common cancer. Scenarios where this does not hold are beyond the scope of this work.
A principal assumption for extrapolation is that the biomarker is equally actionable for both the common and rare cancers, but this might not be the case. Assessment of biomarker actionability in the rare cancer may be informed by considering the two questions outlined below:
Criterion (3a) How strong is the evidence supporting biomarker actionability in rare cancer?
Frameworks ranking biomarkers according to strength of evidence supporting actionability have been published and can inform this assessment in rare cancers.83–89 Top-tier evidence of biomarker actionability for matching targeted therapy is established in prospective, adequately powered RCTs on measures of net clinical benefit—often established for common cancers. In rare cancers where RCTs utilizing these outcome measures are not possible, these frameworks make recommendations for ranking the strength of evidence supporting actionability and include (i) retrospective studies showing clinical benefit from targeted therapy in the biomarker-positive versus biomarker-negative group, (ii) prospective studies showing increased tumor responsiveness without data on survival endpoints, (iii) evidence for a top-tier association but in a different cancer histotype, (iv) preclinical models predicting sensitivity to matched therapy without clinical data, and (v) in silico evidence predicting functional impact similar to that seen for a biomarker-therapy match in different histotypes. Evidence supporting the biological rationale in the rare cancer should be ranked according to strength of clinical validity using these frameworks. Extending these frameworks, we propose considerations for downgrading the strength of the evidence for actionability in the rare cancer below.
Criterion (3b) Is there evidence that suggests the treatment effect in the rare cancer may differ from that in the common cancer thereby not supporting extrapolation?
Clinical, preclinical, and mechanistic evidence for different actionability across cancers should be assessed.
Cellular context and tumor microenvironment
Complex interactions between the biomarker and the cellular context or tumor microenvironment unique to a specific tumor type may exist and alter the actionability of the biomarker. Pembrolizumab received histology-agnostic FDA approval for the treatment of advanced solid tumors with a high tumor mutational burden (TMB-H) for patients who have no other alternative therapeutic options based on the non-randomized, open-label KEYNOTE-158 study. 90 However, clinical benefit was shown to differ across TMB-H tumors where ORR in endometrial cancer was 47% while in anal cancer only 7%, suggesting that tumor microenvironments may influence treatment response and that the predictive ability of TMB may not be uniform across different cancer types.91–94
Compensatory resistance pathways
Even if a molecular alteration is a driver across multiple cancers and these are treated with the same targeted agent(s), emergence of compensatory resistant pathways may differ across cancer types. 95 For example, v-raf murine sarcoma viral oncogene homolog B1 (BRAF) inhibitor vemurafenib has been evaluated in a range of BRAF V600-mutant histotypes including melanoma, NSCLC,96,97 and CRC. 96 In melanoma, RCTs have demonstrated vemurafenib improved OS compared to dacarbazine chemotherapy.98,99 In a non-randomized basket trial, response rates in NSCLC (42%) 96 were comparable to melanoma 99 but no responses were seen in CRC. 96 Subsequent preclinical studies have shown that vemurafenib monotherapy results in rapid acquired resistance to BRAF inhibition in CRC but not in the other cancer types.95,100 This finding has been subsequently confirmed in a prospective RCT with dual inhibition of BRAF and epidermal growth factor receptor pathways. 101
Significant differences in the cellular or tumor microenvironment and/or compensatory resistance pathways between common and rare cancers downgrade the strength of the evidence for actionability and raise uncertainty about extrapolation.
Treatment outcomes
Efficacy
A principal assumption of extrapolation is that the common and rare cancers sharing the same biomarker are similar in prognosis and response to targeted treatment such that the same treatment effect could be expected. 102 When efficacy of targeted therapy is only evaluated in single-arm trials, relative treatment benefit could be extrapolated from the common cancer to the rare cancer provided that: (i) RCT data confirms net clinical benefit in the common cancer, and (ii) signals of efficacy from single-arm or randomized studies in the rare cancer are comparable between the common and rare cancer based on the same validated surrogate endpoint measure(s). 102 Similarly, where clinical benefit of targeted therapy has been demonstrated in a range of heterogeneous cancer types grouped together by the same actionable biomarker profile in a “pan-cancer” study, clinical benefit may reasonably be extrapolated to each rare cancer type provided signals of efficacy on the surrogate measures are similar. For example, fam-trastuzumab deruxtecan is a HER2 directed antibody–drug conjugate which has been shown to improve ORR, duration of response (DOR), and OS compared to physician’s choice chemotherapy in HER2 overexpressed/amplified, previously treated, metastatic breast cancer (ORR 70% vs 29%, median DOR 19.6 months vs 8.3 months, HR for OS 0.66, p = 0.0021). 103 HER2 overexpression is found across diverse cancer types but prevalence rates can be low. In endometrial and cervical cancers, the prevalence of HER2 overexpression in these tumors is approximately 4%. 104 In April 2024, the FDA granted accelerated tumor-agnostic approval to fam-trastuzumab deruxtecan for patients with previously treated, advanced HER2-positive (Immunohistochemistry (IHC) 3+) cancers who have no satisfactory alternative treatment options. 105 This approval was based on a pan-cancer single-arm basket trial showing a comparable ORR of 61.3% and median DOR of 22.1 months. 106 Magnitude of benefit was particularly high in IHC 3+ endometrial (ORR 84.6%, DOR not reached) and cervical cohorts (ORR 75%, DOR 14.2 months). 106 These results compare favorably to historical controls where survival outcomes are poor and chemotherapy response rates are low.107,108 However, there was no observed benefit in the pancreatic cohort (ORR 4%, DOR 5.7 months). 106 Concurrent control comparison may not be feasible in all rare cancer cohorts. Hence uncertainties will remain when the overall treatment effect is applied in each of the different rare cancer cohorts.
Surrogate measures may include progression-free survival (PFS), ORR, pharmacokinetic/pharmacodynamic (PK/PD) properties, circulating tumor DNA levels, and functional imaging responses. Heterogeneity of treatment effect on surrogate measures may be tested. This approach has been supported by regulatory and HTA bodies including the The Food and Drug Administration (FDA), The European Medicines Agency (EMA), United Kingdom’s National Institute for Health and Care Excellence and Australia’s Medical Services Advisory Committee.22,44,61,109
RCTs in rare cancers are ideal wherever possible, including the use of novel trial designs such as randomized basket trials using intermediate outcomes, to strengthen the evidence of efficacy as compared with relying solely on non-randomized trials. Beyond clinical trials in rare cancers, the organized collection of clinical outcome data from post-marketing studies, 110 registries, and real-world studies should be prioritized to continuously build the body of evidence. When considering extrapolation of relative treatment effect, the following questions should be considered:
Criterion (4) Is there a validated surrogate endpoint that can be used to extrapolate the clinical benefit of targeted therapy from the common cancer to the rare cancer? Are estimates of targeted therapy efficacy based on this surrogate endpoint similar between the common and rare cancer?
Treatment outcomes based on surrogate endpoints, such as PFS or ORR, used for extrapolation should be adequately assessed to determine whether they reliably predict treatment benefits for OS.111–114 It is generally not feasible to adequately validate surrogate endpoints in rare cancer studies, particularly in the absence of trials of randomized design. However, these surrogate endpoints should, at a minimum, be validated in the common cancer trials.
Surrogate endpoints are validated for a specified context of use for a specific biomarker, type of therapy, cancer type, and disease setting. Therefore, a validated surrogate endpoint for one cancer type is not necessarily a valid surrogate for a different cancer type. 115 For example, in a study of multiple first-line chemotherapy and hormone therapy trials of advanced cancers, PFS was shown to be an acceptable surrogate for OS in colorectal and ovarian cancers but not in breast and prostate cancers. 116 The minimum size of the surrogate difference or threshold needed to predict a clinical benefit gain (e.g. OS gain) can also differ across cancer types. 117
ORR and disease stabilization measures including DOR and disease control rate (DCR, a combined measure of ORR and stable disease (SD) at a specific time-point) are commonly used as a surrogate for OS in oncology and as a primary endpoint in pivotal trials supporting regulatory approvals in rare cancers.118–120 Tumor shrinkage is regarded to be exceedingly rare in the absence of effective therapy and is widely perceived to precede other clinical improvements, including survival prolongation. However, the validity of ORR as a surrogate for OS has not been established for most settings.121,122 Non-randomized trials have been shown to exaggerate DOR for targeted therapies when compared with RCTs of the same drug for the same setting. 123 DCR as an endpoint also does not completely capture treatment activity as many tumors with indolent natural history will satisfy the criterion of short-term SD. 124
In view of these limitations, alternative endpoints assessing PD response utilizing minimally invasive functional technologies 125 and/or composite endpoints may need to be considered. If validated for the specific context of use, these endpoints may prove useful for extrapolation (Figure 1(d)). Composite endpoints may be particularly useful in rare cancers as they may be more sensitive in detecting the spectrum of treatment effects and reduce sample size requirements.126,127 Composites can also assess more than one aspect of the patient’s health status and incorporate clinically meaningful outcomes. 126 If composite endpoints are used, they should be prespecified, clearly defined, weighted according to clinical relevance, used and reported according to published guidance,128,129 and validated prior to use in different cancer types and clinical settings. Where evidence based on surrogate endpoints does not support similar efficacy between the common and rare cancers, extrapolation may not be appropriate.
Safety
Criterion (5) Are the adverse events experienced in the rare cancer similar to those experienced in the common cancer? Are there any clinically meaningful differences between cancers?
Another important assumption made for extrapolation is that the safety profile of the targeted therapy in rare cancer is similar to that of common cancer. In common cancers, safety data of targeted therapy from RCTs provide an unbiased comparison of adverse events (AEs) related to treatment and differentiate these from disease-related or other non-treatment-related AEs. Safety data is also augmented by post-marketing and real-world studies that capture use in populations outside those highly selected for RCTs.
AEs are likely to be common across multiple cancer types. However, differences can exist across cancer types because of differences in co-existing environmental exposures, comorbidities, organ-specific tumor burden, and prior systemic and local therapies resulting in differing tolerance to treatment-related toxicity.130,131 For example, a meta-analysis of 20 PD-1 inhibitor trials showed significantly higher incidence of pneumonitis in NSCLC and renal cell carcinoma compared to melanoma. 130 Safety data across cancer types should be assessed to judge whether differences are clinically meaningful and are of significant magnitude to represent important high uncertainty, limiting extrapolation. Additional sources that may augment safety data in the rare cancer include natural history studies, auxiliary safety cohorts, expanded access programs, and real-world studies capturing off-label use132,133 and PK/PD data. 125 In controlled “pan-cancer” trials, safety data of the combined control arm may be heterogeneous due to the varying control treatments.
Decision tree
We recommend addressing all criteria for the five components necessary for extrapolation to inform decision-making for the targeted therapy in the rare cancer. Explicit judgments about the level of uncertainty for each component based on an assessment of the supportive evidence will result in a more transparent approach to regulatory decisions. We propose that certainty for all or most of the criteria is required to extrapolate the treatment benefit of targeted therapy from the common to the rare cancer. During the process of evidence evaluation, knowledge gaps may be identified in one or more component(s). Depending on the clinical impact of these gap(s), further research may be needed before extrapolation can be used (Figure 2).
When there is sufficient evidence for provisional or regular regulatory approval, uncertainties may remain regarding the longer-term clinical benefits, safety in broader rare cancer populations, and spectrum of uncommon AEs. Detailed plans for post-approval commitments addressing specific residual uncertainties identified during pre-approval evaluation should be outlined (Figure 2).
Strengths
The framework is an important first step to outline the breadth of criteria essential for evidence assessment for rare biomarker-defined cancers. It is an initial conceptual construct for stimulating multidisciplinary discourse toward developing a validated and reproducible tool that can be incorporated into the HTA process, clinical practice guidelines, and clinical decision-making. Five essential components of evidence assessment from multidisciplinary fields have been incorporated into a single framework. These components should be, but are not commonly, considered as a whole. However, consideration of only one or few components, such as efficacy without addressing prognosis or the analytic validity of the biomarker test, would be incomplete.
Limitations
There are several limitations. The utility of the framework and validity of the approach for judging extrapolation criteria and uncertainty has not yet been assessed. The applicability of the framework across a wide range of targeted therapy-cancer histotype scenarios, as well as reproducibility and consistency of uncertainty judgments require testing.
Future work
As evidence for histology-agnostic-targeted therapies accumulates, the extrapolation criteria may be refined, and anchors developed to guide uncertainty judgments. A transparent process should be developed to assess consistency and reproducibility of uncertainty judgments by independent assessors. This could be undertaken by seeking expert consensus on trialed examples and used to develop a guidance document. Future studies evaluating the utility of this framework for regulatory and reimbursement decisions should be conducted. Outcome measures for these studies may include completeness of evidence assessment and transparency of decisions, time taken from initial targeted therapy approval in a common cancer to additional approvals in other rare cancers sharing the same biomarker, clinical benefit of drugs approved using this framework, and proportion of subsequent withdrawals.
Conclusion
We have proposed a framework for extrapolating evidence of treatment effects for molecularly targeted therapies from common to rare cancers sharing the same predictive biomarker. This framework supports systematic assessment, standardized decision-making, and transparent discussions between key stakeholders. Where there is still insufficient evidence for extrapolation, our approach will also help better target future research to address critical gaps. This will ultimately inform clinical practice and will benefit patients with rare biomarker-defined cancers to access safe and effective targeted therapies.
Supplemental Material
sj-docx-1-tam-10.1177_17588359241273062 – Supplemental material for Criteria for assessing evidence for biomarker-targeted therapies in rare cancers—an extrapolation framework
Supplemental material, sj-docx-1-tam-10.1177_17588359241273062 for Criteria for assessing evidence for biomarker-targeted therapies in rare cancers—an extrapolation framework by Doah Cho, Sarah J. Lord, Robyn Ward, Maarten IJzerman, Andrew Mitchell, David M. Thomas, Saskia Cheyne, Andrew Martin, Rachael L. Morton, John Simes and Chee Khoon Lee in Therapeutic Advances in Medical Oncology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.