
Abstract
Purpose:
This phase 1 study aims to evaluate the tolerability and the recommended phase 2 dose of selinexor in Asian patients with advanced or metastatic malignancies.
Experimental Design:
A total of 105 patients with advanced malignancies were enrolled from two sites in Singapore (National University Hospital and the National Cancer Centre, Singapore) from 24 February 2014 to 14 January 2019. We investigated four dosing schedules of selinexor in a 3 + 3 dose escalation design with an additional Phase 1b expansion cohort. Adverse events were graded with the NCI Common Terminology Criteria for Adverse Events v 4.03. Pharmacodynamic assessments included nuclear cytoplasmic localization of p27, XPO1 cargo proteins pre and post selinexor dosing and pharmacokinetic assessments were conducted at doses between 40 and 60 mg/m2.
Results:
In our Asian patient cohort, dosing at 40 mg/m2 given 2 out of 3 weeks, was the most tolerable for our patients. At this dose level, grade 3 adverse events included fatigue (8%), hyponatremia (23%), vomiting (5%), thrombocytopenia (5%), and anaemia (2%). Selinexor had a rapid oral absorption with median Tmax of 2 h and no PK accumulation after multiple doses of tested regimens. Complete responses were seen in two lymphoma patients. Partial responses were noted in three diffuse large B cell lymphomas, one Hodgkin’s lymphoma and thymic carcinoma patient, respectively.
Conclusion:
Selinexor is tolerated by Asian patients at 40 mg/m2 twice a week given 2 out of 3 weeks. A 1-week drug holiday was needed as our patients could not tolerate the current approved continuous dosing regimens because of persistent grade 3 fatigue, anorexia and hyponatremia.
Background
Selinexor is an oral, slowly reversible, potent and selective inhibitor of nuclear export (SINE) that specifically blocks exportin 1 (XPO1). XPO1, also called chromosome region maintenance protein 1 (CRM1), is the major nuclear export protein in the cell and has found to be overexpressed in many types of cancer. XPO1 (exportin 1) is responsible for the transport of most tumour suppressor and growth regulatory proteins out of the nucleus into the cell. 1 Active nuclear export of these tumour suppressor and growth regulatory proteins is one mechanism malignant cells use to overcome the normal cell cycle and genomic instability checkpoints mediated by these proteins.2,3
To date, selinexor has been approved by the Food and Drug Administration (FDA) for DLBCL (diffuse large B cell lymphoma) patients who have progressed through at least two lines of systemic chemotherapy. The results from the phase 2b SADAL trial of selinexor in relapsed or refractory diffuse large B cell lymphoma showed durable responses and a 13% complete response rate. 4 The combination of selinexor, bortezomib and dexamethasone in the BOSTON trial has also received FDA approval for relapsed or refractory myeloma patients who have received at least one prior line of therapy. 5 The initial phase 2 trial of selinexor in combination with dexamethasone reported an overall partial response rate of 21–26% and median overall survival of 9 months. 6 Subsequent results from the phase 3 BOSTON study which compared once weekly selinexor in combination with weekly bortezomib subcutaneously and low-dose dexamethasone twice-weekly orally to the standard twice-weekly bortezomib plus low-dose dexamethasone showed an improved progression-free survival (PFS) of 13.5 versus 9.5 months (hazard ratio: 0.7; 95% CI: 0.53–0.93; p = 0.0075). 5
The antitumour efficacy of selinexor has also been studied in several phase 1 and 2 solid tumours such as triple negative breast cancer, advanced refractory bone and soft tissue sarcomas, gynaecological cancer and haematological cancers.1,7 –10 The FDA-approved dose for selinexor is at 60 mg orally on days 1 and 3 every week. However, most studies of selinexor have mainly consisted of patients from Western populations and less than 20% of patients were of Asian descent. It is well known that ethnicity can significantly impact on the tolerability, toxicity and efficacy of anticancer agents.11,12 Therefore, this study was conducted with the primary objective of evaluating the safety and tolerability of selinexor in Asian patients and to establish the recommended phase 2 dose (RP2D) in this population of patients.
Methods
This phase 1 study was conducted in accordance with protocol requirements, the International Conference on Harmonization for Good Clinical Practice and the guiding principles in the Declaration of Helsinki, according to local laws and regulations. All enrolled patients provided written informed consent before undergoing study-specific procedures. The protocol was approved by the Domain Specific Review Board (DSRB) in accordance to the International Conference on Harmonization Guidelines for Good Clinical Practice (ICH-GCP), Singapore Guidelines for good clinical practice and all applicable laws and regulations (DSRB approval number 2013/01034, Supplementary Document). This trial is registered under Clinicaltrials.gov with identifier NCT02078349, date of registration was on 5 March 2014.
Eligible patients were recruited from two sites in Singapore (National Cancer Centre, Singapore and National University Cancer Institute, Singapore, National University Hospital). Inclusion criteria required all patients to be at least 21 years of age with histologically documented advanced solid tumours or non-Hodgkin’s lymphoma resistant or refractory to standard treatment; an Eastern Cooperative Oncology Group performance status of 0 or 1; and adequate bone marrow, liver, kidney, coagulation and cardiac function. Exclusion criteria included radiation ⩽ 3 weeks prior to cycle 1 day 1, chemotherapy, or immunotherapy or any other systemic anticancer therapy ⩽ 3 weeks prior to cycle 1 day 1, unstable cardiovascular function, uncontrolled active infection (Hepatitis B and C infection were not exclusion criteria), known HIV infection, renal failure requiring haemodialysis or peritoneal dialysis, pregnancy or breastfeeding concurrent cancer (except nonmelanoma skin cancer or carcinoma in situ of the cervix), unless in complete remission and off all therapy for that disease for a minimum of 3 years, patients with significantly diseased or obstructed gastrointestinal tract, malabsorption, uncontrolled vomiting or diarrhoea or inability to swallow oral medications and unable/unwilling to have a nasogastric or percutaneous endoscopic gastrostomy (PEG) tube inserted. Patients who are unable to swallow may participate if they have a nasogastric or orogastric (NG or OG) or PEG tube inserted to allow administration of the drug in suspension (using Ora-plus) via an oral syringe.
The primary objective was to determine the MTD and recommended phase 2 dose, safety, tolerability and pharmacokinetics of single-agent selinexor (KPT-330) in Asian patients. The RP2D is defined as the next lower dose level below MTD. The MTD is the dose level in which less than one of three patients or greater than or equal to two of six patients experience dose-limiting toxicity (DLT), provided that that dose level is ⩽25% lower than the highest (intolerable) dose tested. The secondary objectives were to determine the pharmacokinetics (PK) and pharmacodynamics (PD) of selinexor and antitumour response in patients with advanced or metastatic solid tumour malignancies. Exploratory objectives included identifying potential predictive biomarkers of relevance.
Study design and treatment
Dose escalation schedules and dose limiting toxicity
In the Phase 1a (dose escalation) phase, we investigated four different dosing schedules [Figure 1(a)]. Schedule 1 was a twice-a-week continuous dose starting at 40 mg/m2 given on Monday and Thursday of every week in a 28-day cycle. This dosing schedule was similar to the RP2D dose obtained in the Princess Margaret cohort that recommended 35 mg/m2 and the current FDA approved dosing of 60 mg twice a week. 9

(a) Treatment schema for phase 1a schedules 1, 2, 3, 4 and phase 1b dose expansion. (b) Consort diagram.
Schedule 2 was a once-weekly dosing starting at 50 mg/m2 given on Monday of every week in a 28-day cycle [Figure 1(a)]. Schedule 3 was a twice-a-week dosing on Mondays and Wednesdays with a 1-week drug holiday and was given for 2 out of 3 weeks in a 21-day cycle [Figure 1(a)]. Schedule 4 was planned to investigate if a lower dose starting at 20 mg/m2, three times a week in a continuous dosing on Monday, Wednesdays and Fridays in a 28-day cycle would be more tolerable [Figure 1(a)].
In the phase 1b expansion cohort, selinexor was dosed at a fixed dose of 60 mg (equivalent of 40 mg/m2) twice a week dosing on days 1, 4, 8 and 11 followed by a 10-day treatment free interval in each 21 day cycle [four doses per cycle, Figure 1(a)]. The rationale for this dosing was concluded after collating our dose escalation data, in discussion with Karyopharm and ongoing pharmacodynamic and pharmacokinetic studies (details given below). Dose level 1 was at 40 mg twice weekly for 2 out of 3 weeks, dose level 2 was at 60 mg once per week for 3 weeks and dose level 3 was at 40 mg once per week for 3 weeks.
Selinexor treatment continued until tumour progression/death, unacceptable toxicity or withdrawal of consent. The MTD was defined as the maximum dose at which the incidence of DLTs during cycle 1 was below 30%. Each cohort was evaluated after patients completed one cycle of treatment or had withdrawn during cycle 1 due to a DLT. DLT was defined as any of the following occurrence in the first cycle at the target dose of the study population that is considered possibly related to the drug. This included grade ⩾ 3 nausea/vomiting, diarrhoea despite taking optimal supportive care and any other grade ⩾ 3 nonhaematological toxicity (except for electrolytes abnormalities that are reversible and asymptomatic or hair loss which is not dose-limiting). Haematological DLT was defined as grade 4 neutropenia [absolute neutrophil count (ANC) < 500/mm3] lasting ⩾ 7 days, febrile neutropenia or grade ⩾ 3 thrombocytopenia associated with bleeding.
Dose escalation followed a modified 3 + 3 design required to establish the MTD. 13 A minimum of three patients were enrolled per cohort. Once three patients are enrolled in a cohort and have completed one cycle of treatment, three additional patients may be added to that cohort. After up to six patients have been accrued to a dose level, that dose level will be closed to accrual until safety assessment of all the three to six patients was performed through a safety cohort meeting at the end of cycle 1. If the dose level was well tolerated at the target dose, then dose escalation was performed in the next cohort of three patients. If none of the patients in this cohort experience DLT during the 4 weeks at the target dose, dose escalation continued as per protocol design.
If one DLT occurred in the first three patients enrolled in a cohort, an additional three patients were enrolled. If another DLT occurred at this dose level (i.e. two DLTs/six patients), this dose will be considered the MTD, and the RP2D defined as the dose level below this dose.
Prophylactic supportive treatment
All patients received prophylactic treatment to prevent anorexia and nausea, which included megestrol acetate 160–400 mg daily, or dexamethasone 2–4 mg on days of dosing and either olanzapine 5.0 mg at bedtime or 2.5 mg twice a day, 0–3 days before the first dosing day of selinexor or mirtazapine 15 mg once a day 0–3 days before the first dose of selinexor.
Study assessment
All adverse events (AE) were recorded and graded using the National Cancer Institute Common Terminology Criteria for Adverse Events v 4.03 (National Cancer Institute 2010) throughout the study period and up to 30 days after the last dose. Toxicity was graded every 2 weeks for the first two cycles and every 4 weeks thereafter, according to National Cancer Institute Common Terminology Criteria for Adverse Events v 4.03. Additional safety evaluations included physical examination, concomitant medications, cardiovascular assessment, vital signs and laboratory assessments which included blood count, clinical chemistry, including liver function test, coagulation and 12-lead electrocardiogram.
Pharmacokinetic assessments
Plasma selinexor levels were collected from 19 individuals in this study who were administered selinexor at doses between 40 mg/m2 and 60 mg/m2 on days 1 and 3 of each week. Six patients at 40 mg/m2 had blood samples for plasma PK collected at: 0, 0.5, 1, 2, 4, 8, 24 and 48 h postdose on cycle 1 day 1. The remaining patients had PK samples collected up to 8 h on cycle 1 day 1. Additional sparse PK samples (0, 0.5, 1, 2 and 4 h postdose) were collected at steady state on cycle 1 day 8, day 15, cycle 2/3 day 1, day 8 or day 15.
Plasma samples were shipped frozen to AIT BioScience, IN, USA, for analysis. Plasma concentrations of selinexor (KPT-330) were determined using a validated liquid chromatography with tandem mass spectrometry (liquid chromatography mass spectrometry) method. The quantification range of selinexor is 1–1000 ng/ml.
Pharmacokinetic parameters were calculated by noncompartmental methodology using Phoenix WinNonlin (Built 8.1.0.3520) software. All plasma concentration values below the lower limit of quantification for the assay were treated as missing in the PK analysis, except for those occurring before the first quantifiable concentration on day 1, which were treated as zero.
Pharmacodynamic assessment
Immunohistochemistry (IHC) studies were performed on tumour samples to determine nuclear localization of XPO1 together with proliferation (Ki67) and apoptotic markers such as ApopTag and cleaved caspase 3. To assess inhibition of XPO1 in peripheral blood mononuclear cells (PBMCs), blood samples were collected before the first dose of selinexor and at 4 and 8 h after the dose. Cells isolated from these samples were evaluated for mRNA expression levels by quantitative reverse transcription–polymerase chain reaction (qRT-PCR) of XPO1, which was upregulated in response to XPO1 inactivation. 14
Efficacy
Tumours were assessed by computed tomography or magnetic resonance imaging for response via Response Evaluation Criteria in Solid Tumours version 1.1, 15 except for patients with lymphoma. For patients with lymphoma, disease response was evaluated by the International Working Group (IWG) Response Criteria for non-Hodgkin’s lymphoma (NHL) 16 and cutaneous T-cell lymphoma (CTCL) consensus response criteria using physical examination, including the Modified Severity Weighted Assessment Tool (mSWAT) 17 for skin assessment, as well as responses in skin, lymph nodes, blood and viscera for CTCL patients. Responses were documented at least once at 8 weeks from baseline and after every two cycles subsequently.
Statistical analyses
All patients who received at least one dose of selinexor were included in the safety evaluation. All patients who completed cycle 1 and received at least one dose during cycle 1 or discontinued during cycle 1 because of a DLT were included in the MTD evaluation. All patients receiving at least one dose of selinexor, and no substantial protocol deviations, were included in pharmacokinetic evaluations; all patients with evaluable pharmacodynamic data, and without substantial protocol deviations, were included in pharmacodynamic evaluations. All patients who received at least one dose of selinexor and had at least one postbaseline tumour scan were included in the evaluation of antitumor activity/response.
Results
Patient demographics and disposition
A total of 105 patients were enrolled from two sites in Singapore (National University Hospital and the National Cancer Centre, Singapore) with the first patient recruited on 24 February 2014 and the last patient recruited on 14 January 2019. Of them, 94 patients received at least one dose of selinexor and 74 patients were on study for at least 28 days [Figure 1(b)]. The median age was 62 years (25–79 years). 61% of patients recruited were men (n = 57) and 39% were women (n = 37, Table 1). 91.5% of the patients were of Asian descent (Chinese, Indian or Malay). 46.9% (n = 45) of patients had four or more lines of prior treatment and all patients had evidence of disease progression prior to enrolment. Eastern Cooperative Oncology Group (ECOG) performance status at baseline was 0 and 1, respectively, in 40% and 60% of patients. The most prevalent cancer types were colorectal cancer (42.5%, n = 40), upper gastrointestinal tract/ hepatobiliary cancers (17%, n = 16) and lymphoma (11%, n = 11), followed by gynaecological cancers (9.6%, n = 9), head and neck/lung cancers (6.4%, n = 6), thymoma and thymic carcinoma (5.3%, n = 5), breast cancer (3.2%, n = 3) and 1 case each for cholangiocarcinoma, tongue squamous cell carcinoma, peripheral nerve sheath tumour, liposarcoma, small cell neuroendocrine tumour and renal clear cell carcinoma (Table 1).

BSA, body surface area; GI, gastrointestinal.
Treatment exposure
In the phase 1a part of the study, all patients were treated with at least one dose of selinexor at dose levels between 16 and 70 mg/m2
In the phase 1b dose expansion cohort, 39 patients received at least one dose of selinexor at the starting dose level of 60 mg twice weekly given 2 weeks on, 1 week off, in a 3-weekly cycle. The median duration of study-drug treatment in the phase 1b dose expansion cohort was 35 days (range: 4–850 days) and median number of cycles was two cycles (range: 1–39 cycles). A total of 11 patients did not receive any study drug treatment and included eight patients who failed the initial screening, one patient who passed away prior to starting study treatment, one patient who was taken off study for splenic abscesses and performance status deterioration prior to starting treatment, and one patient who withdrew from the study prior to the start of treatment. Seven patients withdrew consent after the start of study treatment due to concerns of frequency of visits, the need for a repeat biopsy and intolerance of the study drug.
There were eight deaths that occurred within 30 days of the last study drug administration in the phase 1a cohort, of which five were cancer-related deaths due to progression of disease, two were due to severe pneumonia not related to the study drug and one patient passed away from pulmonary embolism deemed not related to the study drug. Five deaths occurred in the phase 1b expansion cohort, of which four were cancer-related deaths due to progression of disease and one patient passed away from sepsis, not attributed to the study drug.
Safety and tolerability
Dose escalation phase
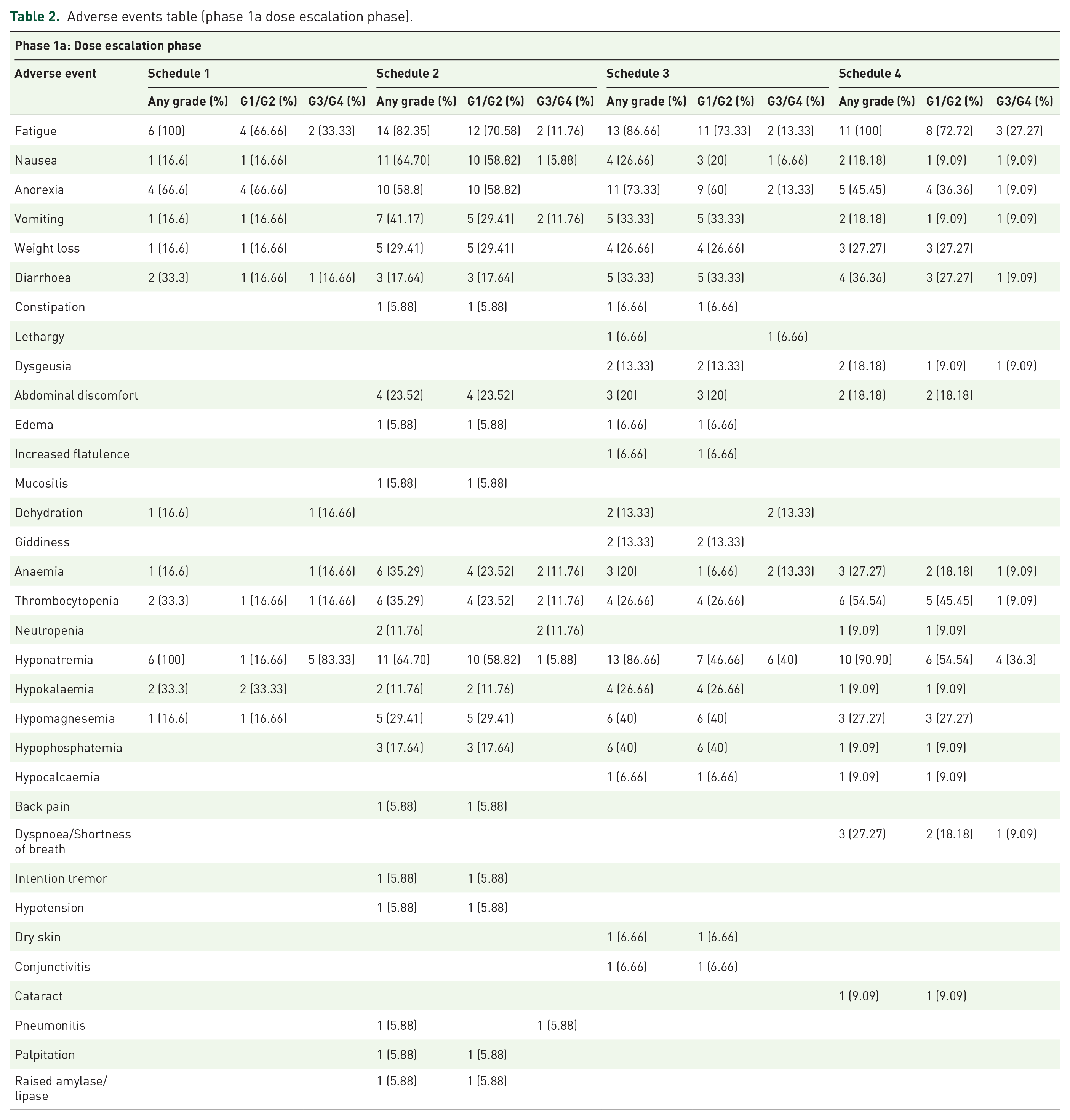
Schedule 1 was stopped after accrual of six patients as although no DLTs were observed, all six patients developed persistent grade 3 fatigue while on the twice-a-week continuous dosing at 40 mg/m2 which resulted in repeated dose delays (Figure 1(a); Table 2).
Adverse events table (phase 1a dose escalation phase).
Schedule 2
At the first dose level of 50 mg/m2 once a week, none of the three patients recruited experienced any DLTs. Dose escalation to 60 mg/m2 in three patients did not show any DLTs. Six patients were recruited at the subsequent dose of 70 mg/m2 with one patient experiencing grade 3 vomiting and one patient experiencing grade 3 thrombocytopenia. An additional four patients were recruited at 70 mg/m2 for safety assessments and one patient experienced grade 3 fatigue that persisted for more than 5 days (Figure 1(a); Table 2).
Given our initial experience with protracted chronic G3 fatigue with continuous weekly dosing regimens, Schedule 3 with a twice-a-week dosing (i.e. with a more than 48 h interval between doses) for 2 out of 3 weeks in a 21-day cycle was subsequently explored to improve tolerability to selinexor. Dosing commenced at 40 mg/m2 with 0/3 patients developing DLTs. The dose was escalated to 50 mg/m2 where six patients were recruited; one out of six patients developed G3 fatigue. Recruitment to this dose level was further expanded to four more patients for safety confirmation. One out of the subsequent four patients developed G3 dehydration from loss of appetite and one other patient developed G3 hyponatremia (Figure 1(a); Table 2). In view of the DLTs experienced at 50 mg/m2, this dose level was declared the MTD and the RP2D was 40 mg/m2 with a schedule of 2 out of 3 weeks in a 21-day cycle.
We further investigated if a lower dose but at increased frequency would be more tolerable in Asians patients. Schedule 4 was three-times-a-week continuous dosing in a 28-day cycle. The dose level started at 20 mg/m2, which recruited six patients (3 + 3), with two out of six patients developing DLTs with G3 fatigue and G3 septicaemia. At dose level 1 at 16 mg/m2, we recruited five patients (3 + 2), of which two out of five patients experienced G3 diarrhoea and G3 syncope (Figure 1(a); Table 2). Hence, this schedule was deemed intolerable and dropped from further development.
Determination of the RP2D
The phase 1a study confirmed our RP2D dose of 40 mg/m2 twice-a-week given 2 out of 3 weeks in a 21-day cycle. Discussions with Karyopharm at this juncture of our clinical trial indicated the company’s plan to eventually move towards an appropriate flat dosing for selinexor rather than a body surface area based dosing. Other phase 1 studies looking at selinexor pharmacokinetics at that time had showed that the interpatient area under curve (AUC) and Cmax variability between body surface area (BSA)-based dosing and flat dosing was comparable. 18 Our pharmacodynamic studies done on patients dosed at 40–70 mg/m2 demonstrated appropriate pharmacodynamic modulation with upregulation of XPO1 even at dose level 40 mg/m2 (detailed results shown in the following). The median BSA of our Asian patients on this trial was 1.5 m 2 , and on discussion with the drug development team, the decision was made to move ahead with a flat dosing of 60 mg per dose, which we deemed appropriate given the mentioned reasons and in addition was consistent with our findings of an RP2D of 40 mg/m2. To confirm the safety of this schedule, 54 patients [Figure 1(b)] were recruited into a dose expansion phase at 60 mg twice a week for 2 out of 3 weeks in a 21-day cycle.
Adverse events in the dose expansion phase dosed at 60 mg twice a week, 2 out of 3 weeks
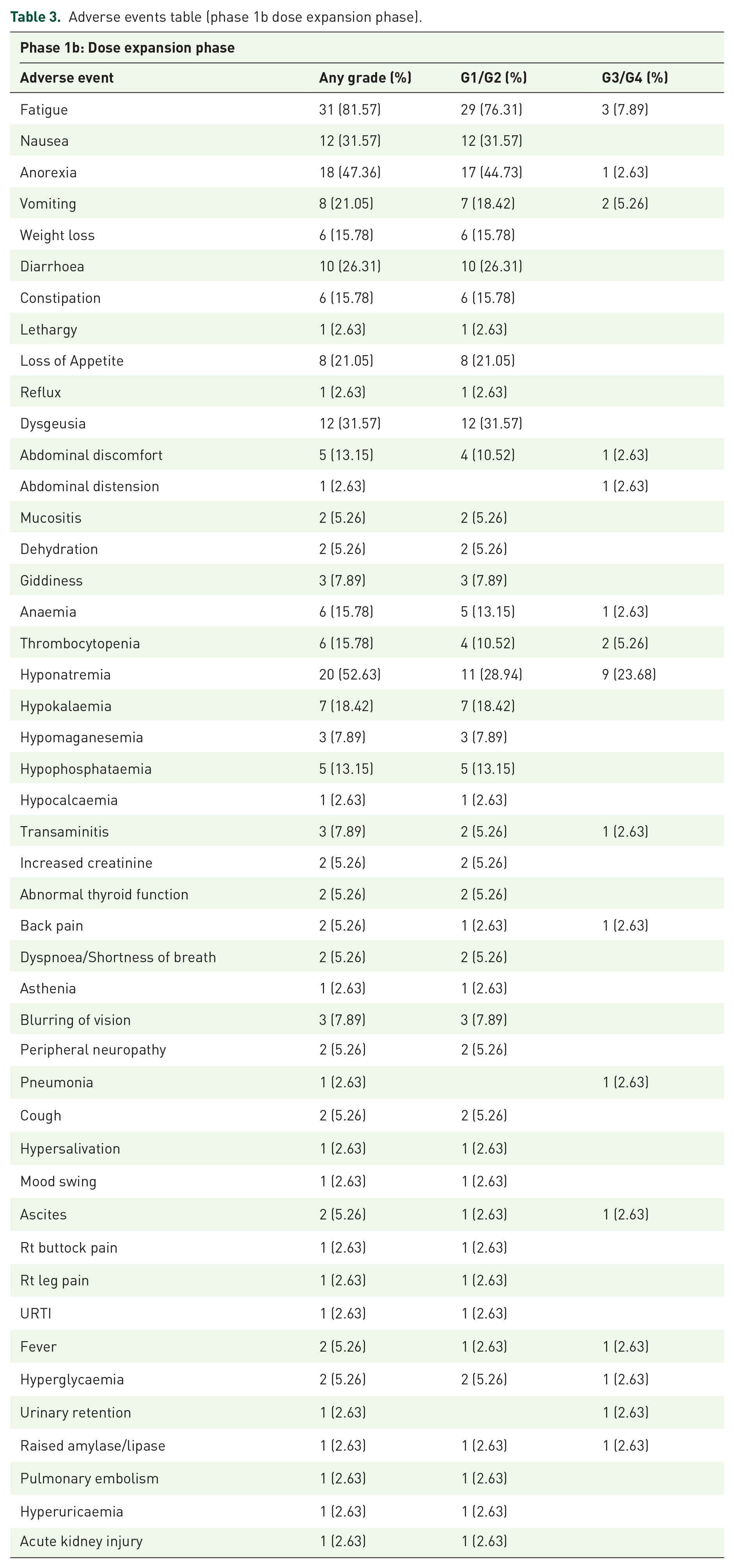
In the dose expansion phase, the most commonly reported AE was fatigue in 80% of our patients (Table 3). The majority of cases were grade 1 or 2. Grade 3 fatigue occurred in 8% of patients. The other common grade 1 or 2 side effects included anorexia (44%), nausea (30%), vomiting (18%), hyponatremia (28%), hypokalaemia (18%), hypophosphatemia (13%) and hypomagnesemia (8%). The most common grade 3 or 4 adverse events were hyponatremia (23%), vomiting (5%), thrombocytopenia (5%) and anaemia (2%). Two patients developed grade 2 elevated transaminases and one patient developed a grade 3 transaminitis which was thought to be related to the study drug. Grade 4 adverse events occurred in two patients in the expansion phase with anaemia and thrombocytopenia. These AE profiles appear consistent with what has been reported in other phase 1/2 human studies involving selinexor; however, the occurrences of hyponatremia and fatigue were higher in our Asian cohort, resulting in the necessity for prophylactic sodium chloride supplementation in the form of sodium chloride tablets and oral rehydration salt sachets. An underlying aetiology for the hyponatremia was not found despite extensive investigation.
Adverse events table (phase 1b dose expansion phase).
Pharmacokinetic and pharmacodynamic analysis
The mean plasma selinexor levels showed a clear separation between the 40 mg/m2 and 60 mg/m2 doses with a delayed peak plasma level in the 50 mg/m2 dose group (Figure 2). The median Tmax values for all dose levels between 40 mg/m2 and 60 mg/m2 at day 1 was 2 h (range: 1–4 h) and the mean T1/2 was 5.8 h (Supplementary Table 1). After multiple doses, the predose concentrations were either 0 or close to 0 on cycle 1 day 15 or cycle 2 day 8/day 15.

(a) Mean ± SD plasma selinexor Concentration versus Time following oral administration of selinexor at 40–60 mg/m2, day 1; (b) mean ± SD selinexor Concentration versus Time following oral administration of selinexor at 40 or 50 mg/m2, days 1 and 8, cycle 1.
The AUC of selinexor between 0 and 8 h (AUC0–8) was dose-proportional among 40, 50 and 60 mg/m2 doses. In the 40 mg/m2 group (n = 6) where PK samples were collected up to 48 h, Cmax was 567 ± 159 (mean ± SD) ng/ml and AUC0–48 was 5345 ± 1541 (mean ± SD) ng*h/ml. These Cmax and AUC values in Asian patients with solid tumour were similar to those observed in Caucasian patients dosed at 39 mg/m2 in a solid tumour Study KCP-330-002 (Cmax = 553 ± 238 ng/ml, AUC0–48 = 5829 ± 1587 ng*h/ml: n = 9). 16 Mean half-life was also similar (~6 h) between this study and study KCP-330-002. 19
IHC analysis was performed on a subset of paired pretreatment and ontreatment tumour biopsies from six patients who consented to biopsies in the initial dose escalation phase (n = 6). In patients with lymphoma, thymoma and colorectal carcinoma, a marked decrease in proliferation and increase in apoptosis were observed in samples after 6 weeks of selinexor treatment as compared with baseline (Supplementary Figure 1). Tumour sections also showed nuclear accumulation of XPO1 cargo protein p53 in a patient with non–small cell lung cancer (NSCLC) (Supplementary Figure 2). To further assess the effects of selinexor on XPO1, we used qRT-PCR to measure XPO1 expression, which is upregulated at the RNA level in response to XPO1 protein inactivation. 13 Of the 10 patients assessed, nine demonstrated at least twofold induction of XPO1 mRNA at 4 h and all 10 patients demonstrated greater than twofold mRNA increases in XPO1 at 8 h at the 40 mg/m2 and 50 mg/m2 doses (Supplementary Figure 3).
In the dose escalation phase 1a study, we observed that RAS- and AKT-pathway-activated colorectal cancer patients appeared to have a longer PFS compared with wild-type tumours (results below, Supplementary Figure 4). Cytoplasmic translocation of p27 could be the key oncogenic mechanism in RAS- and AKT-pathway-activated tumours and potentially targeted by inhibition of XPO1. We commenced screening and recruitment to the Phase 1b part of the study using the above RP2D determined from our Phase 1a trial with a focus on seven prespecified cohorts. These were colorectal cancers with a mutation in the RAS pathway and/or PIK3CA/AKT pathway (n = 17 of which 13 patients had KRAS mutations, two patients had both KRAS and PIK3CA mutations, two patients had NRAS mutations), gynaecological cancers (n = 7), non–small cell lung cancers (n = 1), head and neck or thymic carcinomas (n = 4), any advanced solid malignancy with a mutation in the RAS/PIK3CA/AKT pathway (one non–small cell lung cancer patient with a KRAS G12 C mutation and one ovarian cancer patient with a KRAS G12D mutation), transformed lymphoma or T-cell lymphoma or double expressors diffuse large B cell lymphoma (n = 4) and solid tumours with moderate or high tumour mutational burden less than 10 mutations/mb (none recruited).
Efficacy
Seventy-four patients with solid tumour malignancy treated across all dose levels were evaluated for best response. Confirmed complete responses based on RECIST v1.1 were seen in 2 (2.7%) patients with lymphoma. Duration on treatment of 148 and 1084 days were recorded for both patients respectively. Confirmed partial responses were seen in four (5.4%) patients (two DLBCL, one Hodgkin’s lymphoma and one thymoma). In addition, prolonged stable disease of more than 12 weeks was observed in 10 patients (13.5%). They consisted of six colorectal cancer patients, one pancreatic cancer patient, two thymoma/thymic carcinoma patients and one patient with grey zone lymphoma. Duration of treatment ranged from 42 to 217 days in these patients.
In the phase 1b expansion cohort, 35 heavily pretreated patients with advanced or metastatic malignancy had at least one dose of selinexor and at least one posttreatment scan. In total, 13 (37.1%) patients showed at least stable disease or better as best response according to RECIST v1.1. Duration of treatment ranged from 42 to 217 days in these patients. They included six patients with colorectal cancer, three patients with lymphoma, two patients with thymoma/thymic carcinoma, one patient each had non–small cell lung cancer and ovarian cancer (Figure 3). Among these patients with stable disease, four patients were observed to have a prolonged stable disease of more than 12 weeks. These four patients all had colorectal cancer.
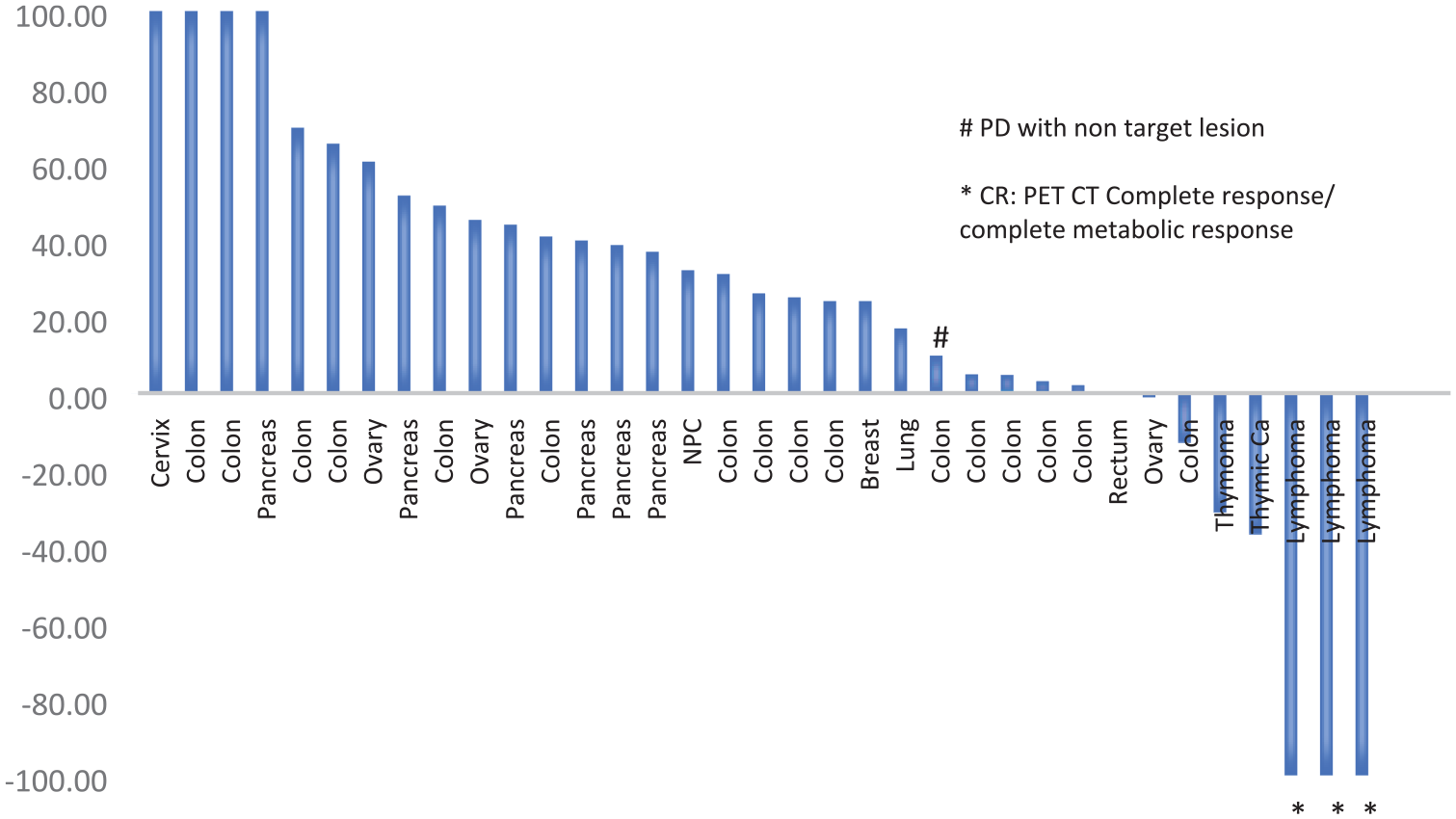
Percentage change in size of target lesions from baseline in evaluable patients in the phase 1b expansion phase.
Subgroup analysis in colorectal cancer patients
Preclinical studies have suggested that activated KRAS and AKT can lead to cytoplasmic localization of the tumour suppressor protein p27, which is a known XPO1 cargo protein, to drive tumour progression.20,21 Given the promising PFS seen in heavily-pretreated KRAS-mutant colorectal cancer patients from the dose escalation study, a prespecified subgroup of colorectal cancer patients with mutations in the RAS and/or PIK3CA/AKT pathway was further recruited in the dose expansion phase 1b study (n = 15). An exploratory subgroup analysis was done on all 36 advanced colorectal cancer patients from both the dose escalation and dose expansion phases. Twenty-six men and 10 women with median age of 65 years (range 48–82 years) were included in this analysis. We observed 10 patients were RAS wild-type, 21 were KRAS mutants, two harboured NRAS mutations, two patients had both KRAS and PIK3CA mutations and one patient had a PIK3CA mutation (Supplementary Table 2). Interestingly, one heavily pretreated colorectal cancer patient with a BRAF V600E somatic mutation, generally recognized to confer a poorer prognosis, had a fairly prolonged PFS of 129 days on selinexor. The median PFS for patients with a RAS/PIK3CA pathway mutation compared with the wild-type patients was 86 versus 50 days (p = 0.09, log-rank), HR: 0.58 (0.25–1.32) (Supplementary Figure 4).
Discussion
Selinexor is a first-in-class inhibitor of nuclear export protein, XPO1, resulting in the accumulation of tumour suppressor proteins within the nucleus and induction of apoptosis in tumour cells. 17 In this phase 1, predominantly Asian cohort (91% Asian descent; 9% others), the most common adverse event was fatigue, which was reported in 80% of our patients. The other more common toxicities were gastrointestinal side effects of anorexia, nausea and vomiting. The spectrum of these toxicities was similar in the predominantly Caucasian cohort (97% Caucasian; 3% Asian) reported by the Princess Margaret group, 9 where up to 70% of patients also reported fatigue as the most common side effects, followed by nausea, anorexia and vomiting. We noticed that our patients as compared with the predominantly Caucasian cohort could not tolerate the continuous dosing regimens because of persistent grade 3 fatigue, anorexia and hyponatremia. Our initial schedule 1, which mirrored the RP2D dose from the Caucasian cohort, was deemed intolerable despite no observable DLTs as all patients developed persistent grade 2 and 3 fatigue beyond cycle 1 (Table 2). In our population which has three main ethnic groups, drug pharmacokinetics did not seem to differ by ethnic populations and hence ethnicity itself would not explain this relative intolerability of continuous dosing we have observed in Asians. One hypothesis is that glutathione transferase metabolism is largely intracellular and may not be reflected in the plasma pharmacokinetic studies. A limiting factor in our study is we did not manage to plan for analyses of genetic polymorphisms where interethnic differences have been described that may account for this difference in drug tolerability that we observed.22,23
Other DLTs included hematologic toxicities of which thrombocytopenia was the most common and occurred earlier as compared with the predominantly Caucasian cohort. 9 In our expansion phase, our recommended RP2D dose was at 60 mg twice a week with a 1-week drug holiday every 3-week cycle. With this schedule and with a pre-emptive anti-emetic schedule, the incidence of grade 3 hyponatremia was 23% and less than 3% of patients had grade 3 or 4 cytopenias. The 1-week drug holiday during each 3-weekly cycle and the addition of a pre-emptive antiemetic regimen of olanzapine or mirtazapine with either megestrol or dexamethasone reduced both the incidence and severity of nausea to 30% and with no grade 3 or more nausea occurring in our patients (Table 3).
We observed an objective response of 8%, where two patients with lymphoma had a confirmed complete response with duration of response more than 5 months. Partial responses were noted in two other cases of DLBCL, one case of Hodgkin’s lymphoma and one thymoma patient (Figure 3). These results are consistent with larger phase 2 studies in DLBCL which the FDA has now given fast track approval for selinexor. 4 In our study, we noted prolonged stable disease, especially in a subgroup of heavily pretreated colorectal cancer patients. RAS mutations in metastatic colorectal cancer confer a known resistance to anti-EGFR agents precluding the use of such drugs in this subgroup of colorectal cancers. Studies and meta-analyses have also suggested that RAS mutations are also a negative prognostic factor with overall poorer survival attributed not just tumour biology but also the lesser available therapeutic options. 24 In view of the data suggesting the RAS/AKT mediated cytoplasmic sequestration of the XPO1 tumour suppressor protein p27 may drive tumour progression, we performed an exploratory analysis looking at the colorectal cancer patients on this study (N = 36) and demonstrated a statistically nonsignificant increase in median PFS for patients with tumours harbouring RAS/PIK3CA pathway mutations compared with patients with RAS/PIK3CA wild-type tumours [86 versus 50 days, p = 0.09, log-rank, HR: 0.58 (0.25–1.32)].
The inherent complexity of the mechanism behind XPO1 inhibition involves the ability of XPO1 to interact with several different tumour suppressors and cell cycle regulators, potentially targeting multiple pathways. An example is the role of XPO1 inhibition leading to the accumulation of TP53 in the nucleus and the impact on tumour suppressor proteins. 25 Despite selinexor’s known impact on tumour suppressor proteins, a study in KRAS-mutant NSCLC, using mainly TP53 mutant cell lines, differed in their selinexor sensitivity, 26 suggesting that nucleocytoplasmic localization of other XPO1-associated tumour suppressor cargos, such as p27, in a specific oncogene-activated context, may have a role in determining the responses to selinexor in solid tumour malignancies. Studies involving larger cohorts of patients with these potential biomarkers will need to be performed before any meaningful conclusions can be drawn.
Selinexor monotherapy has shown limited potential in several clinical trials for the treatment of solid tumour malignancy. A phase 1 study of 189 patients with advanced solid tumours evaluated the safety and efficacy of selinexor. 9 In total, 157 patients were deemed evaluable and only one complete and six partial responses were observed (ORR 4%). In addition, several phase 2 trials further tested single-agent selinexor for the treatment of specific solid tumours with varying disappointing outcomes. In a phase 2 study of 14 patients with metastatic castration-resistant prostate cancer, monotherapy selinexor resulted in 50% reduction in PSA values of only two patients. 25% of patients (two of the eight) with measurable disease at baseline achieved partial response and 50% of patients (four of the eight) achieved stable disease as their best radiographic response. 27 A phase 2 trial was also conducted using single-agent selinexor in 10 heavily pretreated patients with metastatic triple-negative breast cancer (NCT02402764). Disappointingly, monotherapy selinexor did not result in any objective responses. 7 However, our preliminary data demonstrating improved PFS for KRAS/AKT mutant colorectal cancer, albeit statistically nonsignificant, does provide a hint towards the need for incorporation of better biomarker selection to identify patients most likely to benefit from selinexor monotherapy in future studies. Recently, selinexor has been shown to increase PD-1 and CTLA4 gene expression in leukocytes and induced PD-L1 gene expression in human melanoma cell lines suggesting that a combination with immune checkpoint inhibitors may be a viable strategy. 28 One such study is the NEXUS study combining selinexor with ipilimumab and nivolumab (NCT04850755). Several other studies are also exploring combinations with chemotherapy and other novel agents including ixazomib (a proteasome inhibitor) in advanced sarcoma (NCT03880123), paclitaxel and carboplatin in advanced ovarian and endometrial cancers (NCT02269293), gemcitabine and nab-paclitaxel in pancreatic cancer (NCT02178436) 29 .
In conclusion, our study shows that selinexor is well tolerated by Asian patients at a dose of 60 mg twice a week, but given 2 out of 3 weeks with a 1-week drug holiday, which is contrary to the current approved continuous dosing with monotherapy. As monotherapy, our study of heavily pretreated patients with advance/ metastatic malignancy showed a confirmed complete and partial response rate of 8% with a prolonged stable disease of more than 12 weeks in 13% of patients in this study. We envisage that the RP2D identified in this study can serve as the backbone for future selinexor monotherapy and drug combination studies in biomarker selected cohorts that may contain a significant population of Asian patients.
Supplemental Material
sj-docx-1-tam-10.1177_17588359221087555 – Supplemental material for A phase 1 study of the safety, pharmacokinetics and pharmacodynamics of escalating doses followed by dose expansion of the selective inhibitor of nuclear export (SINE) selinexor in Asian patients with advanced or metastatic malignancies
Supplemental material, sj-docx-1-tam-10.1177_17588359221087555 for A phase 1 study of the safety, pharmacokinetics and pharmacodynamics of escalating doses followed by dose expansion of the selective inhibitor of nuclear export (SINE) selinexor in Asian patients with advanced or metastatic malignancies by Jingshan Ho, Valerie Heong, Wei Peng Yong, Ross Soo, Cheng Ean Chee, Andrea Wong, Raghav Sundar, Yee Liang Thian, Anil Gopinathan, Mei Yan Pang, Priscillia Koe, Santhiay Nathan Jeraj, Phyu Pyar Soe, Mu Yar Soe, Tiffany Tang, Matthew C.H. Ng, David W.M. Tai, Tira J.Y. Tan, Hongmei Xu, Hua Chang, Yosef Landesman, Jatin Shah, Sharon Shacham, Soo Chin Lee, Daniel S.W. Tan, Boon Cher Goh and David S.P. Tan in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-2-tam-10.1177_17588359221087555 – Supplemental material for A phase 1 study of the safety, pharmacokinetics and pharmacodynamics of escalating doses followed by dose expansion of the selective inhibitor of nuclear export (SINE) selinexor in Asian patients with advanced or metastatic malignancies
Supplemental material, sj-docx-2-tam-10.1177_17588359221087555 for A phase 1 study of the safety, pharmacokinetics and pharmacodynamics of escalating doses followed by dose expansion of the selective inhibitor of nuclear export (SINE) selinexor in Asian patients with advanced or metastatic malignancies by Jingshan Ho, Valerie Heong, Wei Peng Yong, Ross Soo, Cheng Ean Chee, Andrea Wong, Raghav Sundar, Yee Liang Thian, Anil Gopinathan, Mei Yan Pang, Priscillia Koe, Santhiay Nathan Jeraj, Phyu Pyar Soe, Mu Yar Soe, Tiffany Tang, Matthew C.H. Ng, David W.M. Tai, Tira J.Y. Tan, Hongmei Xu, Hua Chang, Yosef Landesman, Jatin Shah, Sharon Shacham, Soo Chin Lee, Daniel S.W. Tan, Boon Cher Goh and David S.P. Tan in Therapeutic Advances in Medical Oncology
Footnotes
Author contributions
Conflict of interest statement
The author declared the following potential conflicts of interest with respect to the research, authorship and/or publication of this article: D.S.P.T. received consultancy fees from AstraZeneca, Roche, MSD, Merck Serono, Tessa Therapeutics, Eisai and Genmab; D.S.P.T. received research funding from AstraZeneca, Bayer and Karyopharm. V.H. received consultancy fees for AstraZeneca, Pfizer, DKSH and Novartis; Stock Ownership: Asian Microbiome Library (AMiLi); the sponsor had no involvement in the design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review or approval of the manuscript; or the decision to submit the manuscript for publication.
Funding
The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study was supported by grant funding from Karyopharm, Pangetsu Family Foundation Gynaecological Cancer Research Fund and the National Medical Research Council Singapore, National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative. D.S.P.T. was supported by the Singapore Ministry of Health’s National Medical Research Council Transition Award (NMRC/TA/0019/2013) and Clinician Scientist Award (NMR/CSA-INV/0016/2017) and Pangestu Family Foundation Gynaecological Cancer Research Fund. V.H. was supported by the Yong Loo Lin fellowship grant and the NHG-LKC Medicine Clinician Scientist Career Scheme grant (CSCS/20001).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.