
Abstract
Background:
Immune checkpoint inhibitor therapy has demonstrated impressive clinical benefits in multiple tumor types. TQB2450, a novel monoclonal antibody targeting programmed cell death ligand 1, has shown safety and efficacy in preclinical studies.
Objectives:
This first-in-human study aimed to evaluate the safety/tolerability, pharmacokinetics (PK), immunogenicity, and preliminary antitumor activity of TQB2450 in patients with advanced malignant tumors.
Design and methods:
In this phase I study, eligible patients with advanced malignant tumors received intravenous TQB2450 once every 3 weeks. This study consisted of a 3 + 3 dose-escalation phase (1–30 mg/kg) and a specific dose-expansion phase (1200 mg). The primary endpoints were maximum tolerated dose (MTD), dose-limiting toxicity (DLT), and safety. The secondary endpoints were PK, immunogenicity, and investigator-assessed response rate.
Results:
Between April 2018 and February 2020, 40 patients were enrolled (22 in the dose-escalation phase and 18 in the dose-expansion phase). No DLT was reported and the MTD was not reached. Grade ⩾3 or worse treatment-related treatment-emergent adverse events (AEs) occurred in 11 (27.50%) patients, with the most frequent being aspartate aminotransferase increased (5.00%), leukopenia (5.00%), and anemia (5.00%). Treatment-related serious AEs were reported in six patients, the most common of which was decompensated liver function (5.00%). No treatment-related death was reported. The maximum serum concentration of TQB2450 increased in a dose-proportional manner. Treatment-induced anti-drug antibodies were detected in 31.58% (12/38) of patients. The investigator assessed the objective response rate as 5.00% and the disease control rate was 52.50%, including 2 partial responses and 19 stable diseases. The median progression-free survival was 2.69 (95% confidence interval, 2.07–6.14) months.
Conclusion:
TQB2450 has a manageable safety profile with favorable PK and immunogenicity and has shown early evidence of clinical activity in advanced malignant tumors.
ClinicalTrials.gov identifier:
NCT03460457.
Introduction
Immunotherapy, which is developed based on tumor immune escape mechanisms, aims to boost natural defenses to eliminate malignant cells.1,2 In recent years, immunotherapy has revolutionized the treatment landscape for multiple hematologic and solid malignancies.1,3 Programmed cell death 1 (PD-1) is an inhibitory immune receptor predominantly expressed on the surface of activated T and B lymphocytes,4,5 whereas its ligand programmed cell death ligand 1 (PD-L1) is commonly overexpressed on the surface of tumors and immune cells in the tumor microenvironment. 6 The PD-1/PD-L1 interaction in the tumor microenvironment offers an immune escape mechanism for tumor cells. 7
In recent years, immunotherapies that inhibit the immune checkpoint interaction between PD-1 and PD-L1 have shown substantial survival benefits across a wide spectrum of tumors.8,9 Notably, PD-L1 inhibitors appear to be associated with a lower mean incidence of grade 3 or worse adverse events (AEs) compared to PD-1 inhibitors,10,11 suggesting potential clinical advantages of PD-L1 inhibitors. Based on the immense success in clinical trials, three PD-L1 inhibitors, including atezolizumab, durvalumab, and avelumab, have been approved by the Food and Drug Administration (FDA) for the treatment of various types of cancer. 12 Unfortunately, despite the promising clinical activity of PD-L1 inhibitors in cancer treatment, a low response rate of patients, primary or acquired immune resistance, and immune-related AEs, have limited their clinical application.11,13,14 Therefore, it is urgent to develop novel PD-L1 inhibitors to meet the growing clinical need for efficacy and safety.
TQB2450 is a novel humanized immunoglobulin G1 (IgG1) monoclonal antibody against PD-L1. 15 In the cellular assay, the TQB2450 effectively blocked the interaction of PD-L1 with PD-1 and the binding of PD-L1 with CD80, and strongly activated T cells by the production of interferon-gamma in a mixed lymphocyte reaction. 16 Besides, the potent antitumor activity of TQB2450 was confirmed in mouse models of melanoma and colon cancer. 16 Additionally, TQB2450 showed pharmacological activity and was well tolerated with a wide margin of safety, which was well demonstrated in preclinical pharmacodynamic and toxicological studies. 16 Based on these preclinical results of TQB2450, this first-in-human phase I trial was conducted to evaluate the safety and tolerability based on dose-limiting toxicities (DLTs) and maximum tolerated dose (MTD), determine pharmacokinetics (PK) and immunogenicity profiles, and to explore the preliminary antitumor activity of TQB2450 in patients with advanced malignant tumors.
Methods
Study design and patients
This was an open-label, single-arm, dose-escalation/dose-expansion, phase I study of TQB2450 in patients with advanced malignant tumors. Patients were eligible if they were aged 18–70 years with histologically/cytologically confirmed advanced malignant tumors that had disease progressed or developed intolerable toxicity to standard treatment. Eligible patients had an estimated life expectancy of at least 3 months, an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0–1, and adequate organ function. Patients in the dose-expansion phase had predominantly gastrointestinal tumors, such as gastric, bowel, and esophageal cancer, with at least one measurable tumor lesion according to Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1), without suitable chemotherapy regimen, or unwillingness to be treated with chemotherapy.
Key exclusion criteria for both study phases included any prior PD-1 or PD-L1 inhibitors treatment, prior systemic treatment with glucocorticoids or other immunosuppressive agents within 4 weeks before enrollment, severe allergy to other monoclonal antibodies drugs, major surgery within 4 weeks before the first administration of TQB2450 or the presence of unhealed wounds, ulcers, or fractures, active or history of autoimmune disease, or uncontrolled systemic disease, except for radiation-induced local interstitial pneumonia. Full eligibility and exclusion criteria are provided in Supplemental Appendix 1.
This trial is registered with ClinicalTrials. gov, NCT03460457. Additionally, the reporting of this study conforms to the Strengthening the Reporting of Observational Studies in Epidemiology statement. 17
Procedures
All patients received intravenous TQB2450 (a humanized IgG1 PD-L1 inhibitor provided by Chia Tai Tianqing Pharmaceutical Group Co., Ltd, Jiangsu, China) once every 3 weeks. In the dose-escalation phase, the dose level (1, 3, 10, 20, and 30 mg/kg) was escalated according to a 3 + 3 design with each cohort starting with three patients [except for the first dose cohort (n = 1)]. The decision to proceed to the next dose level in this phase was made based on the incidence of DLT during the first 21-day treatment cycle. If a DLT was reported in one of three patients, then up to three additional patients were treated at the same dose level. If no additional DLT occurred, the next dose level was initiated. If a second patient in a cohort had a DLT, study treatment in that cohort was to be stopped. DLTs were defined as any grade 4 hematologic AEs or any grade ⩾3 non-hematologic AEs (excluding controlled nausea, vomiting, and diarrhea). Of note, for patients who present with grade 2 aspartate aminotransferase (AST) and/or alkaline phosphatase increased at baseline, their liver function parameters elevated to >10 × upper limit of normal (ULN) were also considered as DLT.
During the dose-expansion phase, patients with advanced malignant tumors received TQB2450 1200 mg (recommended phase 2 dose, RP2D) intravenously once every 3 weeks. For both phases, treatment was continued until disease progression, unacceptable toxicity, withdrawal of consent, or at investigator’s discretion. Dose reductions or delays of TQB2450 were permitted in case of unacceptable treatment-related toxicity. Criteria for dose suspension and treatment discontinuation are described in Supplemental Appendix 1. Additionally, treatment would be continued when patients experienced initial disease progression and met the criteria for continuation of treatment, following discussions between the investigators and the sponsor (Supplemental Appendix 1).
Tumor response was assessed by the investigator per RECIST 1.1 or Lugano 2014 criteria (hematologic malignancies), using magnetic resonance imaging or computed tomography at baseline, every three cycles until cycle 18, and followed by every four cycles thereafter during treatment. Patients were continuously monitored for safety and tolerability during the study. All patients received TQB2450, therefore, all AEs, regardless of causality, were considered treatment-emergent AEs (TEAEs). All TEAEs were evaluated and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03.
Endpoints
The primary endpoints were the assessment of the safety/tolerability and identification of the MTD and DLT. The main safety endpoints were the incidence of TEAEs, treatment-related TEAEs, serious AEs (SAEs), and AEs of special interest (AESIs). MTD was defined as the highest drug dosage not expected to lead to DLT in >33% of patients during the first 21 days.
The secondary endpoints included PK, immunogenicity, and preliminary antitumor activity. Antitumor activity endpoints were progression-free survival (PFS), objective response rate (ORR), and disease control rate (DCR). PFS was defined as the time from the start of treatment to disease progression or death from any cause, whichever occurred first. ORR was defined as the proportion of patients with the best tumor response as complete response (CR) or partial response (PR). DCR was defined as the proportion of patients with the best tumor response of CR, PR, and stable disease (SD).
PK and immunogenicity
Blood samples were collected at baseline and at scheduled time points after a dose of TQB2450 for the determination of PK parameters and immunogenicity analysis. PK parameters, including maximum serum concentration (Cmax), terminal half-life (T1/2λ), the volume of distribution (Vd), clearance (CL), and area under the concentration–time curve (AUC), were estimated using Phoenix WinNonlin version 6.4 (Pharsight Corporation, Mountain View, CA, USA) with a non-compartmental model. The antibody to TQB2450 [anti-drug antibody (ADA)] was assessed by characterizing the total ADA concentrations. Confirmed-positive samples would be further characterized by titration and with a neutralizing antibody assay. Detailed information on the sampling schedule and analytical methods for PK and immunogenicity are presented in Supplemental Appendix 1.
Statistical analysis
The sample size of the dose-escalation phase was determined by the observed toxicities according to the 3 + 3 design with three–six patients in each dose cohort. For dose-expansion, sample sizes were determined based on the clinical, empirical, and practical considerations used for phase I studies, without statistical considerations. A maximum of 80 patients were planned to be enrolled in this phase to observe the safety or efficacy signals.
The safety analysis set (SS) included patients who received at least one dose of TQB2450. Patients who received at least one dose of TQB2450 and had at least one post–treatment serum concentration of TQB2450 were evaluated for PK and immunogenicity analysis. The antitumor activity analysis was performed for patients in the full analysis set (FAS), which was defined as all patients who received at least one dose of TQB2450 and had at least one post-baseline efficacy assessment. Descriptive statistics included means with standard deviations or medians with minimum and maximum for continuous variables and counts and percentages for categorical variables. PFS was analyzed using the Kaplan–Meier method and expressed as median values with corresponding two-sided 95% confidence intervals (CIs). For the analysis of PFS, data for patients who were alive and without disease progression or lost to follow-up at the time of the last imaging assessment were censored. SAS software (SAS Institute Inc., Cary, North Carolina, USA) was used for all statistical analyses.
Results
Baseline characteristics
Between April 2018 and September 2019, 53 patients were screened for enrollment, of which 13 were excluded for failure to meet eligibility criteria (n = 11), withdraw consent (n = 1), and judged as ineligible at the investigator’s discretion (n = 1). Finally, 40 patients with advanced malignant tumors were enrolled in the dose-escalation phase (n = 22, 1 mg/kg cohort, n = 1, 3, 10, and 30 mg/kg cohorts, n = 6 each, 20 mg/kg cohort, n = 3), and the dose-expansion phase (n = 18, 1200 mg cohort, Figure 1). At the time of data cut-off (31 December 2021), all 40 patients were included in the SS and FAS, and 39 patients were included in the PK and immunogenicity analysis set.
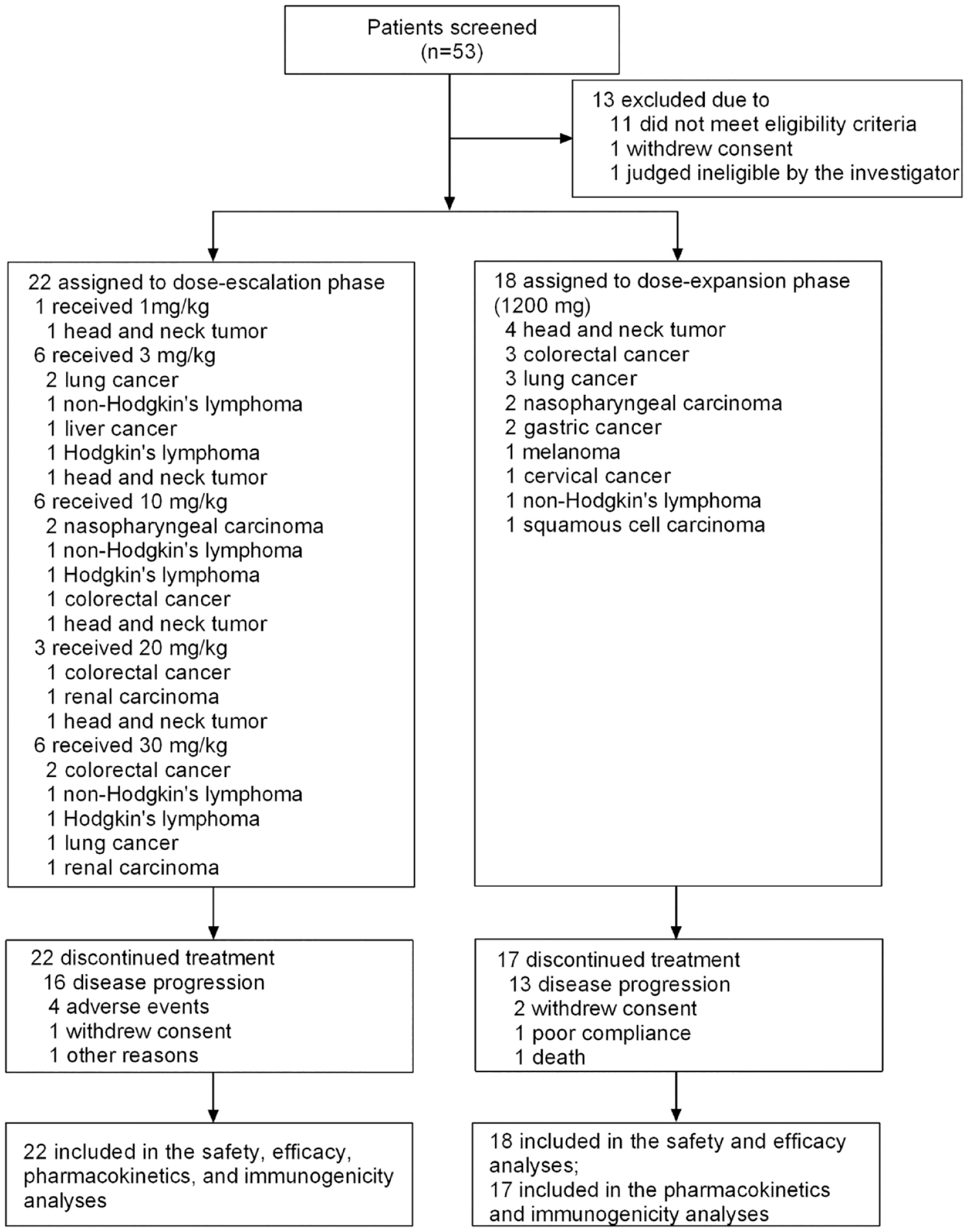
Trial profile.
Demographic and baseline characteristics of the patients are presented in Table 1. Patients had a variety of tumor types, including head and neck tumors (n = 8), colorectal cancer (n = 7), lung cancer (n = 6), non-Hodgkin’s lymphoma (n = 4), nasopharyngeal carcinoma (n = 4), Hodgkin’s lymphoma (n = 3), gastric cancer (n = 2), renal carcinoma (n = 2), liver cancer (n = 1), cervical cancer (n = 1), melanoma (n = 1), and squamous cell carcinoma (n = 1). The median age of the enrolled patients was 54 years (range, 30–69), of these, 75.00% were male. A total of 92.50% of patients had a baseline ECOG PS of 1. All patients were heavily pretreated at study enrollment, with 90.00% receiving chemotherapy, 67.50% surgery, 60.00% radiotherapy, 35.00% targeted drugs or immunotherapy, and 37.50% other anti-tumor therapies. Eighty-five percent of the included patients had concomitant/medical histories, of which 94.44% were in the 1200 mg cohort.
Demographic and clinical characteristic of the patients at baseline during dose-escalation and dose-expansion.
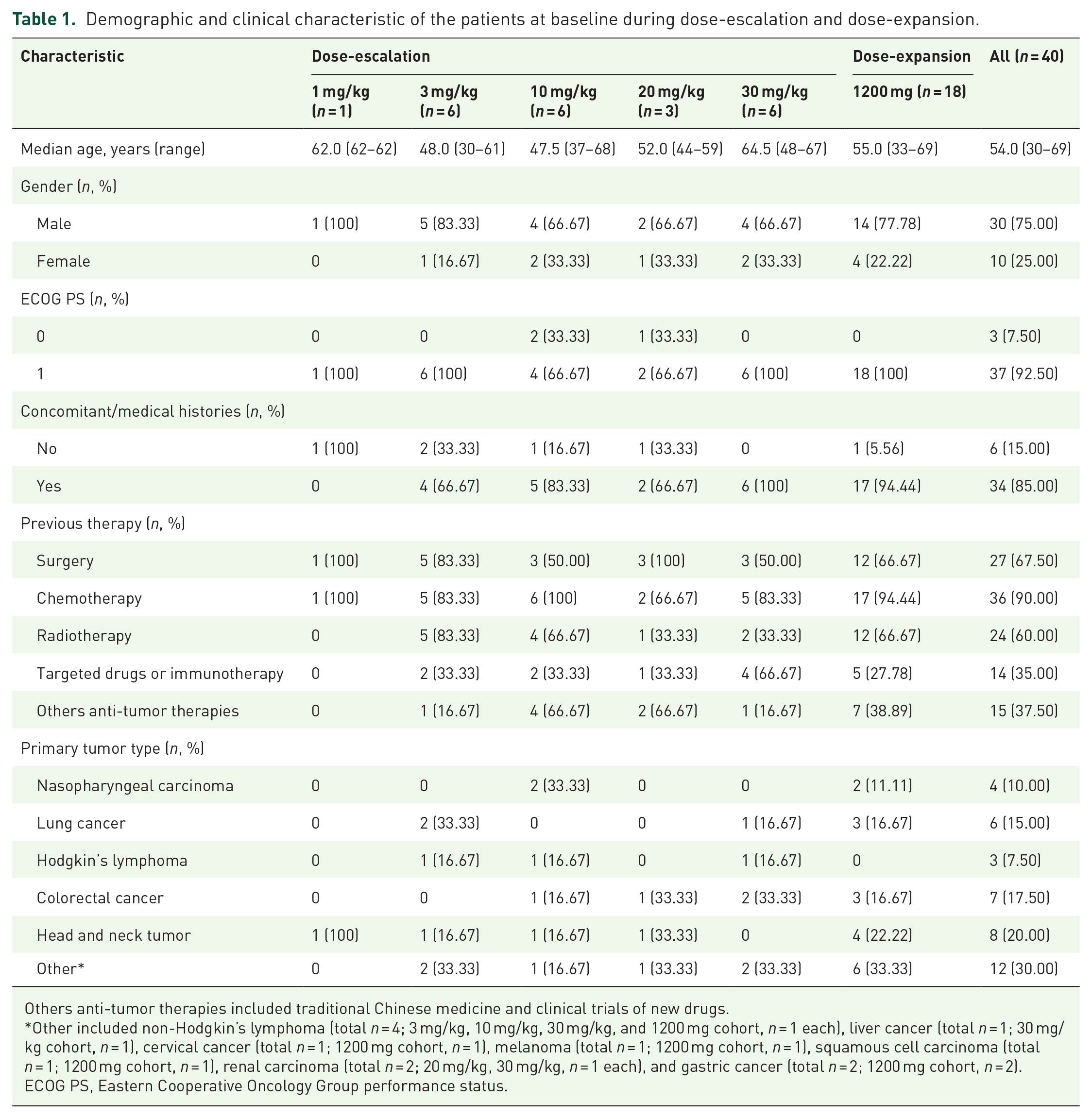
Others anti-tumor therapies included traditional Chinese medicine and clinical trials of new drugs.
Other included non-Hodgkin’s lymphoma (total n = 4; 3 mg/kg, 10 mg/kg, 30 mg/kg, and 1200 mg cohort, n = 1 each), liver cancer (total n = 1; 30 mg/kg cohort, n = 1), cervical cancer (total n = 1; 1200 mg cohort, n = 1), melanoma (total n = 1; 1200 mg cohort, n = 1), squamous cell carcinoma (total n = 1; 1200 mg cohort, n = 1), renal carcinoma (total n = 2; 20 mg/kg, 30 mg/kg, n = 1 each), and gastric cancer (total n = 2; 1200 mg cohort, n = 2).
ECOG PS, Eastern Cooperative Oncology Group performance status.
DLT, MTD, and RP2D
Among patients eligible for DLT assessment (n = 22) during the dose-escalation phase, no DLT was observed in any cohort. Consequently, the protocol-defined MTD was not reached (NR). It should be noted that the 3, 10, and 30 mg/kg cohorts were each expanded to six patients for blood sample collection. The RP2D of TQB2450 was determined as 1200 mg once every 3 weeks on the basis of previous preclinical data, receptor occupancy (RO) and immunogenicity of TQB2450, and the recommended dosage of other PD-L1 inhibitors.
Safety and tolerability
The median duration of treatment was four cycles (range, 1–40), with 87.50% (35/40) of patients receiving more than three cycles of TQB2450. The median relative dose intensity of TQB2450 in this study was 100% (range, 100–102%). All 40 patients who received at least one dose of TQB2450 were included in the SS. A summary of TEAEs in all grades and grade ⩾3 in the safety population is shown in Table 2. Forty patients experienced at least one TEAE of any grade, with grade 3 or worse occurring in 18 (18/40, 45.00%) patients. Treatment-related TEAEs were observed in 38 (95.00%) patients, of which 11 (27.50%) were reported as grade 3 or worse (3 of 18 patients at 1200 mg cohort). The most common treatment-related TEAEs of any grade were AST increased (12/40, 30.00%), γ-glutamyltransferase (γ-GT) increased (9/40, 22.50%), thyroid-stimulating hormone increased (9/40, 22.50%), hyperuricemia (9/40, 22.50%), and anemia (9/40, 22.50%). AST increased (2/40, 5.00%), leukopenia (2/40, 5.00%), and anemia (2/40, 5.00%) were the most frequent grade 3 or worse treatment-related TEAEs.
TEAEs in safety population (n = 40).

AESI, adverse events of special interest; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ECG, electrocardiogram; SAE, serious adverse effects; TEAE, treatment-emergent adverse event.
SAEs occurred in 11 (27.50%) patients. Of these, six patients developed a treatment-related SAE, including decompensated liver function (n = 2), hypercalcemia (n = 1), lethargy (n = 1), pleural effusion (n = 1), and febrile neutropenia (n = 1). Notably, no treatment-related SAEs occurred in the 1200 mg cohort. All patients experienced at least one AESI, of which 29 (72.50%) reported a treatment-related AESI. Additionally, none of the patients receiving 1200 mg of TQB2450 reported grade 3 or worse treatment-related AESI. Across the entire study, no treatment-related deaths were reported. A total of seven (7/40, 17.50%) patients (two patients at 1200 mg cohort) experienced treatment interruptions (7/40, 17.50%) due to TEAEs, of which five (one patient at 1200 mg cohort) were considered as treatment-related.
PK outcomes
Group mean estimated PK parameters using non-compartmental analysis from 39 patients are listed by cohort in Table 3. The observed mean TQB2450 concentration–time profiles for all dose groups are shown in Figure 2.
Summary of PK parameters for TQB2450 (n = 39).

AUC0–t, area under the plasma concentration–time curve from time 0 to the time of the last quantifiable concentration; AUC0–∞, area under the plasma concentration–time curve from time 0 to infinity; CL, clearance; Cmax, maximum observed plasma concentration; PK, pharmacokinetics; SD, standard deviations; Tmax, median time to peak plasma concentration; Vd, volume of distribution.
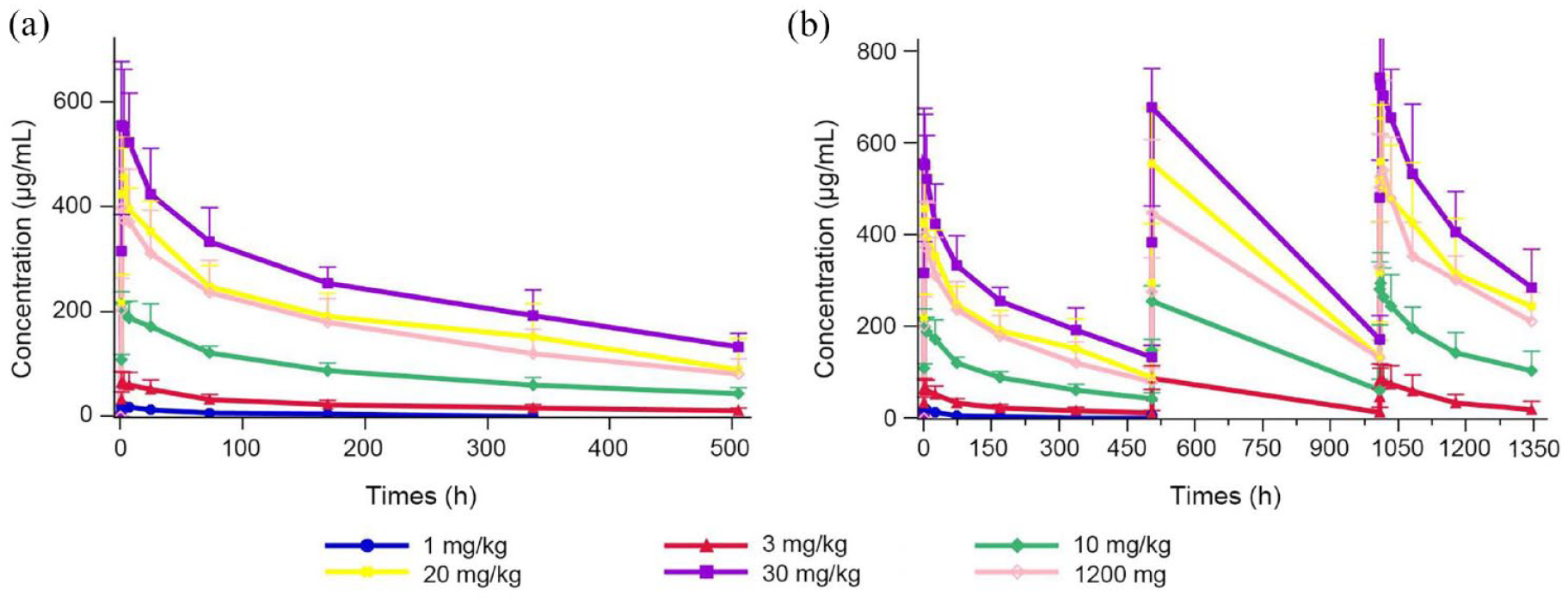
TQB2450 serum concentration versus time profiles for all dose groups. (a) First treatment cycle. (b) All treatment cycles and follow-up.
After administration of TQB2450, the Cmax increased in a dose-proportional manner across the dose range of 1–30 mg/kg; however, mean estimates of AUC0–t and AUC0–∞ were increased greater than dose-proportionally manner. TQB2450 was rapidly absorbed following administration (Figure 2), the peak concentrations of all doses occurred at 3.00 h, except for the 10 mg/kg dose at 1.31 h. The average apparent terminal half-life of TQB2450 ranged from 3.55 to 14.36 days. The Vd maintained relative stability across dose levels (except for the 1 mg/kg dose).
Immunogenicity
A total of 39 patients were tested for immunogenicity (Table 4). Of the 39 patients, one patient was ADA-positive at baseline. Treatment-induced ADAs were detected in 12 of 38 patients who were ADA-negative at baseline. The incidence of immunogenicity varied across dose levels (Table 4). For the 12 patients who developed ADAs following treatment, the median time to the first detection was 22.0 ± 21.19 h and the median duration of ADA positivity was 85.20 ± 68.35 h. Of the 12 patients with treatment-induced ADA, 5 were tested negative for ADA at the last assessment.
Antidrug antibody.

SD, standard deviations.
Antitumor activity
At the data cut-off (31 December 2021), the median follow-up duration was 27.30 months [95% CI, not reached (NR)-NR]. In the overall population, 2 patients (5.00%) achieved a PR [3 mg/kg (Hodgkin’s lymphoma) and 1200 mg cohort (lung cancer), one each], 19 patients (47.50%) experienced SD (9 at 1200 mg cohort), 14 patients (35.00%) had PD (6 at 1200 mg cohort) (Table 5), and 5 were not evaluable. The ORR and the DCR were 5.00% (95% CI, 0.87–18.21%) and 52.50% (95% CI, 36.34–68.18%), respectively. Figure 3 provides additional detail regarding the depth of response and duration of the treatment. At the time of the study cut-off, eight patients had received more than 6 months of therapy, with one patient remaining on treatment after 1 year of initial dosing [Figure 3(a)]. The median PFS was 2.69 months (95% CI, 2.07–6.14, Figure 4).
Antitumor activity of TQB2450.

95% CI, confidence interval; CR, complete response; DCR, disease control rate; NE, not evaluable; ORR, objective response rates; PD, progressive disease, PR, partial response; SD, stable disease.

Tumor response. (a) Time to response and duration of response. Each bar represents one patient. (b) Waterfall plot of maximum percent change in tumor size of solid tumors (n = 29) from baseline as measured by Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1). (c) Waterfall plot of maximum percent change in tumor size of hematologic malignancies (n = 5) from baseline as measured by Lugano 2014 criteria. (d) Percentage change in tumor size of solid tumors (n = 29) from baseline as measured by RECIST 1.1. (e) Percentage change in tumor size of hematologic malignancies (n = 5) from baseline as measured by Lugano 2014 criteria.

Kaplan–Meier curve of progression-free survival.
Discussion
This first-in-human dose-escalation/dose-expansion phase I study evaluated TQB2450 (a novel PD-L1 inhibitor) in patients with advanced malignant tumors. No patients in the dose-escalation phase experienced a protocol-defined DLT; therefore, the MTD of TQB2450 was NR. Overall, TQB2450 showed a manageable safety profile with no unexpected safety signals and a lower incidence of treatment-related SAEs observed during the study. In addition, TQB2450 showed preliminary evidence of antitumor activity in this population with an encouraging DCR of 52.50% and a median PFS of 2.69 months (95% CI, 2.07–6.14). These findings supported further studies regarding the safety and antitumor activity of TQB2450 in specific malignancies enrolled in our study.
The RP2D of TQB2450 in our study was determined as 1200 mg once every 3 weeks on the basis of previously unpublished preclinical data, RO and immunogenicity of TQB2450, and the recommended dosage of other PD-L1 inhibitors. The previous vitro peripheral blood mononuclear cell (PBMC) RO assay indicated that TQB2450 at 0.0625 μg/mL achieved RO >90% of PBMC from different donors. PK/PD results from a preclinical trial with colon MC-38/H-11 mouse model showed a tumor-to-blood ratio of 0.013 for TQB2450. In combination with the RO assay, a TQB2450 blood concentration of at least 4.8 μg/mL was required to ensure a drug concentration of 0.0625 μg/mL in tumor tissue, therefore requiring a minimum TQB2450 plasma trough concentration (Ctrough) of 4.8 μg/mL for all patients in the phase II study. The dose of 3 mg/kg once every 3 weeks was excluded based on phase I clinical PK results, which showed that several blood concentrations in subjects at this dose were below 4.8 μg/mL after cycle 3 due to ADA formation. The study found that all six patients at 10 mg/kg once every 3 weeks cohort had a Ctrough level of greater than 4.8 μg/mL and meaningful pharmacodynamic effects. Therefore, the recommended clinical dose was at least 10 mg/kg once every 3 weeks. In addition, when establishing the RP2D, we also took into consideration the dosages of other PD-L1 inhibitors, such as atezolizumab (1200 mg once every 3 weeks), sugemalimab (1200 mg once every 3 weeks), envafolimab (150 mg once week), avelumab (10 mg/kg once every 2 weeks), and durvalumab (10 mg/kg once every 2 weeks). Taking into account that the occurrence of ADA at a low dose level could prevent reaching the required blood concentrations for RO (>90%) in vitro, whereas the occurrence of ADA at a higher dose level had little impact on drug exposure. Therefore, under the premise of at least 10 mg/kg once every 3 weeks, it is advisable to select the higher possible dose whenever possible. Considering the average body weight of Chinese patients, RO, and immunogenicity of TQB2450, as well as the dose, efficacy, and safety of other anti-PD-L1 antibodies, the RP2D of TQB2450 was determined as 1200 mg once every 3 weeks.
In the present study, the safety profile of TQB2450 was generally tolerable and manageable in patients with advanced malignant tumors. Generally, the majority of treatment-related TEAEs were regarded as grade 1–2, and only a few treatment-related grade 3–4 TEAEs (27.50%) or SAEs (15.0%) were reported in our study, which was comparable to those of other PD-L1 inhibitors, with approximately 13.3–35.0% reporting grade 3–4 TEAEs18,19 and 11.8–24.5% reporting SAE.19 –21 The most common treatment-related TEAEs were AST increased, γ-GT increased, thyroid-stimulating hormone increased, hyperuricemia, and anemia, which were consistent with the safety profile of previous reports of TQB245022 –24 or other PD-L1 inhibitors.25 –27 Importantly, the majority of AEs were generally mild, tolerable, and reversible. Five patients with dose interruption had been recovered from treatment-related TEAEs by supportive care. Moreover, no unexpected TEAEs or treatment-related deaths were observed in the present study. The reported immune-related AEs were only alanine aminotransferase increased and AST increased, without colitis, hypothyroidism, pneumonitis, or rash, which were commonly reported with other PD-L1 inhibitors in a meta-analysis. 11 Nevertheless, the results should be interpreted prudently due to the small sample size. Consistently, as reported in several studies, the addition of TQB2450 to anlotinib monotherapy did not show any new safety signals of anlotinib in various tumors including non-small cell lung cancer and esophageal squamous cell carcinoma.28 –33 Taken together, TQB2450 could be considered safe and tolerable for patients with advanced malignant tumors as regards all these data.
TQB2450 exhibited a favorable PK profile, with the Cmax increased in a dose-proportional manner. It was rapidly absorbed after dosing, and the peak concentrations of all doses occurred at ⩽3.0 h. The Vd of TQB2450 (approximately 7 L) was similar to those reported for the PD-L1 inhibitor atezolizumab (6.9 L), 34 higher than durvalumab (5.6 L) 35 and avelumab (4.72 L). 36 Besides, the average terminal half-life of TQB2450 ranged from 3.55 to 14.36 days, which was comparable to that observed for envafolimab (7–23 days) 37 and avelumab (6.1 days). 36 Moreover, the CL of TQB2450 (approximately 0.4 L/day) was also consistent with the data reported from other PD-L1 inhibitors (0.12–0.59 L/day).34 –36 However, all these results should be interpreted carefully due to the high variability and small sample size. As the first published PK data for TQB2450 in patients with advanced malignant tumors, it would be interesting to compare our PK findings with subsequent PK data that may emerge from the ongoing clinical trials.
In terms of immunogenicity, treatment-induced ADAs were detected in 12 (31.58%) patients who were ADA-negative at baseline. This result was consistent with the data from atezolizumab (13.1–54.1%) 38 but higher than other PD-L1 inhibitors such as avelumab (4.1%) and durvalumab (2.9%). 39 Due to the differences in patient baseline characteristics, cancer types, sample sizes, and ADA detection methods across different studies, it is crucial to compare ADA incidence rates for different PD-L1 inhibitors with caution. Additionally, the duration of the ADA effect is an important indicator of the relevant clinical impact of ADA.40,41 In the present study, 5 of 12 patients who tested ADA positive during treatment turned negative at the last assessment, with one patient having a duration of ADA response for only 41 days. These early transient occurrences of ADA may be primary immunoglobulin M (IgM) responses rather than long-lasting IgG responses, with IgM responses less likely to lead to detrimental clinical impact. 42 However, these results should be confirmed in further investigation.
Preliminary clinical activity was observed in enrolled patients, with 2 PR and 19 SD. Despite our ORR (5.00%) was slightly lower than other PD-L1 antibodies (8.0–11.8%),21,26,43,44 TQB2450 demonstrated a more favorable DCR (52.50%) versus envafolimab (34.30%) 44 and LY3300054 (33.30%) 43 in a similar population. Significantly, our study had fewer patients with immune-sensitive cancers (7.5%) and with an ECOG PS of 0 (7.5%) compared to avelumab (23.5%, 82.4%) 21 and LY3300054 (39.1%, 56.5%). 43 Moreover, since all patients here were unscreened for PD-L1, a small proportion with high PD-L1 expression cannot be ruled out. These factors might explain our slightly lower ORR data. Besides, our PFS data (2.69 months) was comparable to the results for atezolizumab (2.8 months) 45 and MEDI4736 (approximately 2 months). 46 Overall, the preliminary activity of TQB2450 supports further investigation with a large sample size.
A noteworthy finding of our study was the durable response obtained following TQB2450 treatment in two patients, including lung cancer (25.26 months) and Hodgkin’s lymphoma (6.27 months). In lung cancer, our data (ORR, 16.7%, DCR, 50.0%) resembled historical results for TQB2450 alone/with anlotinib (ORR, 25.0%, DCR, 100.0%) 32 or other PD-L1 inhibitors (ORR, 14.0–21.0%, DCR, 24.0–64.0%) in early-phase studies.47 –49 PD-L1 overexpression in hematological malignancies indicated potential for PD-1/PD-L1 inhibitors, 50 but data on PD-L1 inhibitors for Hodgkin’s lymphoma were scarce.27,51,52 Interestingly, our low-dose TQB2450 monotherapy (3 mg/kg) achieved an ORR of 33.3% in Hodgkin’s lymphoma, similar to other PD-L1 inhibitors (22.22–41.9%).27,52 This result underscored TQB2450’s potential in treating Hodgkin’s lymphoma, warranting future research.
The present study had several limitations. As our study was an early-phase clinical trial, the antitumor activity of TQB2450 in specific tumor types had not been adequately explored. Meanwhile, the results of our study should be viewed as exploratory, with results to be confirmed in ongoing, tumor-type-specific, larger clinical trials. Additionally, analyses of biomarkers, such as PD-L1, infiltrating T cells, or other biomarkers as potentially valuable biomarkers should be considered to determine associations with response to TQB2450 in future investigations. Finally, given that our study was conducted only in a Chinese population, the safety, tolerability, and antitumor activity of TQB2450 need to be further investigated in the Caucasian population.
Conclusion
TQB2450 is a novel PD-L1 inhibitor that was well tolerated in patients with advanced malignant tumors. Common AEs were manageable with treatment interruptions or supportive care. Favorable PK and immunogenicity of TQB2450 were also observed. Additionally, TQB2450 demonstrated antitumor activity with encouraging DCR in this population. These findings suggested that TQB2450 might be a potential treatment option for advanced malignant tumors. Based on these findings from the phase I study of TQB2450, several ongoing phase II and phase III trials of TQB2450 have been conducted for more than 2 years including non-small cell lung cancer (NCT04964479), endometrial cancer (NCT04574284), and kidney cancer (NCT04523272). Furthermore, results from the phase III trial in small cell lung cancer (NCT04234607) have been disclosed at the World Lung Cancer Congress. The combination of TQB2450, anlotinib, and chemotherapy demonstrated significant benefits compared to placebo and chemotherapy in terms of median PFS (6.9 versus 4.2 months), median OS (19.3 months versus 11.9 months), and ORR (81.3% versus 66.8%).
Supplemental Material
sj-docx-1-tam-10.1177_17588359231220516 – Supplemental material for TQB2450 in patients with advanced malignant tumors: results from a phase I dose-escalation and expansion study
Supplemental material, sj-docx-1-tam-10.1177_17588359231220516 for TQB2450 in patients with advanced malignant tumors: results from a phase I dose-escalation and expansion study by Junli Xue, Liqiong Xue, Wenbo Tang, Xiaoxiao Ge, Wei Zhao, Qun Li, Wei Peng, Congqi Dai, Ye Guo and Jin Li in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
The authors thank the clinical investigators and associated staff of clinical operations for their contributions to this work, as well as the patients and their families for their participation in this research. The authors also thank Chia Tai Tianqing Pharmaceutical Group Co., Ltd for contributions to the drugs and funding for this study.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.