
Abstract
Objectives:
Clinical trials are complicated, expensive, time-consuming, and frequently do not lead to discoveries that improve the health of patients with disease. Adaptive clinical trials have emerged as a methodology to provide more flexibility in design elements to better answer scientific questions regarding whether new treatments are efficacious. Limited observational data exist that describe the complex process of designing adaptive clinical trials. To address these issues, the Adaptive Designs Accelerating Promising Treatments Into Trials project developed six, tailored, flexible, adaptive, phase-III clinical trials for neurological emergencies, and investigators prospectively monitored and observed the processes. The objective of this work is to describe the adaptive design development process, the final design, and the current status of the adaptive trial designs that were developed.
Methods:
To observe and reflect upon the trial development process, we employed a rich, mixed methods evaluation that combined quantitative data from visual analog scale to assess attitudes about adaptive trials, along with in-depth qualitative data about the development process gathered from observations.
Results:
The Adaptive Designs Accelerating Promising Treatments Into Trials team developed six adaptive clinical trial designs. Across the six designs, 53 attitude surveys were completed at baseline and after the trial planning process completed. Compared to baseline, the participants believed significantly more strongly that the adaptive designs would be accepted by National Institutes of Health review panels and non-researcher clinicians. In addition, after the trial planning process, the participants more strongly believed that the adaptive design would meet the scientific and medical goals of the studies.
Conclusion:
Introducing the adaptive design at early conceptualization proved critical to successful adoption and implementation of that trial. Involving key stakeholders from several scientific domains early in the process appears to be associated with improved attitudes towards adaptive designs over the life cycle of clinical trial development.
Keywords
Introduction
Clinical trials provide a rigorous methodology that remains a standard for developing and testing treatments. However, clinical trials are complicated, expensive, time-consuming, and frequently do not lead to discoveries that improve the health of patients with disease. 1 Even extremely effective new treatments have an overall development time-scale usually measured in decades. Many potential explanatory factors account for the long duration of clinical translation from bench to bedside. 2 The very structure of clinical trials can account for both the high failure rate of new treatments and the lengthy discovery process. 3 The structure of trials, along with the other issues slowing discovery, were recognized as an area for further research and innovation by the US Food and Drug Administration (FDA) and the National Institutes of Health (NIH). 4 Together, they launched the Advancing Regulatory Science initiative with the goal of learning about new ways to speed discoveries that can safely and effectively improve human health.
Clinical trial development in the US public sector
The US NIH is composed of several, mostly disease-specific institutes and centers, for example, the National Institutes of Neurological Disorders and Stroke (NINDS) focuses on the clinical neurosciences. The NIH funds foundational science (molecular biology, genetics, pathophysiology), translational science (includes animal studies that inform how well treatments work), and clinical research. Clinical trials are a subset of the latter category where patients are prospectively enrolled in scientific experiments to test how treatments work. Such treatments include drugs, devices, and behavioral interventions. Most commonly, university-based researchers submit research proposals to the NIH. These proposals provide a relatively short description of the planned science. Then, external, independent scientists peer review and prioritize the proposals based on potential impact, validity of scientific approach, and other factors such as the track record of the investigative team. Funding for the planning processes to develop a complete clinical trial design and reproducible scientific protocol is frequently non-existent or quite limited.
How investigator-initiated, NIH-directed trials are different from industry projects
Generally speaking, drug and device manufacturers are for-profit entities. As such, they have specific obligations to owners and shareholders to develop products that will be efficacious and profitable. Clinical trial development is actively funded by these firms. This includes the biostatistical design of trials and the development of detailed clinical trial protocols and procedure manuals. Similar to NIH-funded trials, a main barrier to successful trials in this space is successful recruitment, enrollment, and follow-up of eligible patients. The primary hurdle after that is regulatory approval. If more complicated designs that require pre-trial mathematical simulation are deemed necessary by the trial development teams, the process to invest resources to accomplish that is relatively straightforward and quick. This contrasts with the NIH grant submission, review, and funding decision process which typically has a duration of at least 2 years.
How publically funded clinical trials in the United States differ from other countries
As noted above, most clinical trials funded by the NIH are investigator initiated, and the resources for planning prior to a grant submission are limited. Since clinical trials are complicated endeavors, many logistical, biostatistical, and scientific issues pertaining to clinical trials are addressed in limited fashion prior to the submission of a grant proposal. Some important exceptions exist. The NINDS requires that any large-scale clinical trial involving a drug or device must first be approved or exempted by the FDA. The submission to this regulatory process requires a detailed protocol and background information. This presents an additional barrier to investigators who may not have time or resources at their institution to achieve this submission. An additional challenge is getting additional help from other key scientists such as ethicists, research coordinators, or biostatisticians to get an FDA submission ready for a grant that may never be funded. In contrast, the United Kingdom funds several clinical trial methodology hubs. These hubs do not have funds to conduct research protocols but provide a professional class of scientists who have both the time and the expertise to carefully develop clinical trial designs and protocols.
How clinical trials tend to differ from bench research
Typically, institutions make investments in laboratories and equipment for scientists who engage in non-clinical research. When research proposals are submitted in this space, the basic tools to conduct the research often exist. This contrasts with a clinical trial, where there is usually no dedicated infrastructure to screen, enroll, and follow patients. In addition, scientists in non-clinical research can often start preliminary experiments in support of grant applications using resources from existing funding. In contrast, it is generally not feasible to start clinical trials without dedicated financial and scientific resources.
The Adaptive Designs Accelerating Promising Treatments Into Trials project
The Adaptive Designs Accelerating Promising Treatments Into Trials (ADAPT-IT) project was one of the four cooperative awards funded as part of Advancing Regulatory Science. 5 Briefly, an adaptive clinical trial (ACT) is a clinical experiment that uses information accruing from patients as the trial is conducted to inform future trial decisions in a pre-specified, algorithm-driven way. 6 For example, a study evaluating five doses of a new treatment may start to allocate more patients to the dose that appears to be benefiting patients the most, ideally in a way that maintains blinding of the enrolling investigators. In addition to blinding, adaptive elements are pre-specified to reduce bias and potential validity threats. The goals of ADAPT-IT were to develop four, tailored, flexible, adaptive, phase-III clinical trials for neurological emergencies and to use a rich mixed methods approach to observe and reflect upon the trial development process. The laboratory for ADAPT-IT was the Neurological Emergencies Treatment Trials (NETT) network. 7 NETT is funded by the NINDS. Trials are proposed by principal investigators (PIs) and must undergo peer review at NINDS. NETT is administered by both a Clinical Coordinating Center (CCC) and a Statistics and Data Management Center (SDMC).
Understanding ACTs in the academic setting
As an academic project, ADAPT-IT largely focused on ACTs from a scientific and clinical perspective, as opposed to industry-driven projects which tend to have more focus on efficiency and cost-effectiveness. 8 ADAPT-IT provided funding for trial design work, which has not typically been the model for NIH-funded clinical trials. Projects designed using the ADAPT-IT model still needed to go through usual NIH procedures for approval to submit large-scale grants with budgets over $1 million USD per year. After that, they also proceeded through peer review and if successful through these steps needed to be approved by an institute’s advisory council (similar to a board of directors). Outside that model, academic investigators generally must develop a protocol and grant proposal without specific funding—this typically limits the amount of time and resources that could be allocated towards detailed pre-trial design simulations or other activities essential to planning more complicated designs.
Standard NETT clinical trial development procedure
Generally, the procedure for developing clinical trials in NETT prior to ADAPT-IT was as follows. Areas of interest were identified by investigators, both inside and outside of the network, and discussed with key scientists at NETT and NINDS. Usually, phase-I or II trial was previously conducted prior to these discussions. Often these exploratory trials used a variety of different patient populations, treatment windows, and possibly different doses and durations of the same treatment (drugs or devices). Since direct comparison was not usually possible, a consensus protocol for the phase-III trial was developed by expert opinion. Several key decisions regarding the above trial operating characteristics (who and when to enroll, what and how much to use) were made, usually with limited comparative quantitative information (particularly on dose and schedule). The design was summarized in a grant and concurrently submitted to the FDA, if needed, for Investigational New Drug (IND) or Investigational Device Exemption (IDE) review.
ADAPT-IT development process
In contrast, the design process implemented in ADAPT-IT focused on identifying and discussing the areas of uncertainty that residually existed when moving into the phase-III design (as opposed to minimizing them and making strong assumptions from the limited phase-II data). 9 The collaborative team of biostatisticians, pre-clinical scientists, and clinicians had early discussions to prioritize areas of important uncertainty and create a trial concept that learned about these An example area is potentially narrowing a therapeutic window from 6 to 4 h based on a pre-specified rule if patient outcome data suggested futility within the later time window, as opposed to finishing an overall futile trial that enrolled the bulk of its patients between 6 and 4 h.
Previous ADAPT-IT findings
Several important observations from the process evaluation of ADAPT-IT have already emerged. Our baseline mixed methods evaluation identified substantial variations in attitudes and beliefs among clinical trial stakeholders, regarding greater use of adaptive designs in confirmatory trials. 10 We additionally focused on the participant’s perceptions regarding potential ethical advantages and disadvantages to adaptive trials. 10 We found an ethical advantage was that the higher number of patients who received a better performing treatment in one particular scenario within one type of an adaptive trial (response adaptive randomization). However, concerns regarding patients and researchers potentially altering their enrollment patterns was an area of ethical concern, as this could compromise scientific validity. One potential concern is waiting for more information to accrue within a trial before deciding to enroll a patient, although the NETT focus on unpredictable emergencies made this unlikely for the trial ideas used in ADAPT-IT. In a more holistic process evaluation, we observed that the participants became more willing to consider flexible designs as our project progressed. 10 In addition, we observed a large need for education of clinicians and biostatisticians alike. Clinicians needed a better understanding of what the potential benefits of an adaptive design could be, tempered by what realistically could be done. Biostatisticians needed to develop better methods to communicate complicated designs efficiently and succinctly.
With the current understanding of potential barriers to adaptive designs, along with our preliminary understanding of how the process of developing trials within ADAPT-IT functioned, it is important to consider how each of the individual trials progressed. In addition, the methodology by which the NETT network tended to alter its trial development procedures was important to catalog and describe cumulatively. The objective of this work is to describe the adaptive design development process, the final design, and the current status of six different adaptive trial designs that were developed.
Methods
Study design
We conducted the evaluation using a longitudinal case study, mixed methods research design using field observations, and a 21-item ACT beliefs survey with visual analog scale (VAS). The VAS matched similar themes as the baseline attitudes survey, but asked participants for opinions regarding the current clinical trial they were designing. Additional data sources were summaries of the meetings, querying of trial PIs for current status, and textual analysis of summary statements from grant submissions.
Settings and participants
Participants were recruited as part of the ADAPT-IT project, exploring the incorporation of ACT designs into an existing NETT network.5,7 Project investigators held a series of meetings that included experts in ACT design and investigators interested in developing an ACT for specific research topics related to neurological emergencies. A mixed methods team evaluated the ACT development process during these meetings and conducted the analysis. While the initial aim of ADAPT-IT was to develop four trials, the team was able to design six distinct trials during the study period.
Data collection
Data were collected via field self-administered VAS surveys (see Online Appendix), direct queries of PIs for status reports, summary statements provided from the NIH grant review process, and observations of trial development meetings by the mixed methods team (M.F., S.M., L.L.) through writing extensive field notes. Observations are one form of qualitative data widely used in the social sciences and often underutilized in the health sciences. In the study, they were particularly useful for understanding the behaviors that occurred during the meetings. The observations were led by two researchers trained in qualitative research, including the team leader through medical anthropology (M.F.) and the other through her dissertation (L.L.). Observational data were collected by in-person, participant-observation at all meetings, meaning the observers interacted some during the meeting. Demographic information was collected from the participants using the same instrument as our prior work. 11 Data were collected between January 2011 and August 2015. Participants were classified as belonging to one of the following groups of clinical trial experts: academic clinician researchers (n = 23), academic biostatisticians from NIH-funded clinical trial networks with substantial experience running phase-III trials (n = 18), consultant biostatisticians working in academic or industry settings with specific experience in Bayesian adaptive designs (n = 13), and other stakeholders, for example, NIH officials, FDA statisticians, medical officers, and patient advocates—all experts in the planning of clinical trials (n = 18).
Variables
Participants considered advantages and disadvantages from the perspectives of the patient, researcher, FDA, and NIH. Survey questions were formulated to gather opinions of the clinical trial experts regarding the ethical advantages and disadvantages of ACT designs. Additionally, the progress and current status of the clinical trial proposals was summarized by direct contact with each of the trial PIs (or lead investigator preparing the grant proposal).
Data sources
As previously described in detail, participants answered the VAS items by completing a paper survey or a web-based survey. 11 The VAS allowed participants to mark a point of agreement on a continuum ranging from “definitely not,” to “probably not,” to “possibly,” to “probably,” to “definitely.” We used a 100-point scale to allow more resolution to examine differences than a five-point structured Likert-type scale would allow, as we desired respondents to make estimations on a probability scale. To compute a quantitative measure of a participant’s assessment, we assigned the lowest anchor a value of 0, and the highest anchor a value of 100, and calculated a level of agreement score based on the point chosen by the participants for the VAS items. The VAS was offered right after the first face-to-face meeting, and again just after the final face-to-face meeting for each trial. Trial PIs also provided the narrative summary statements from grant submissions of the designs.
Data analysis
Descriptive statistics (proportions and means) were calculated for demographic variables. The VAS data were summarized by trial and by type of stakeholder. The mean and standard deviation for the final stakeholder attitudes were calculated. In addition, the mean difference from responses on the final survey versus the baseline survey was calculated with 95% confidence interval (CI). The instruments are fully described and available for download in our previous report, limited to the ethical aspects of this part of the mixed methods evaluation of ADAPT-IT (provided in Online Appendix). 11 Narrative field notes of trial development progress were maintained by the mixed methods team and summarized over time. After each meeting, the observations were discussed in order to draw among the qualitative team and the entire ADAPT-IT team after each meeting when field observations were collected. A team of qualitative experts (author 1, author 2) coded and analyzed the field notes. The summary statements were reviewed by the mixed methods team and also briefly summarized. In addition, themes from the summary statements were inductively derived and presented. These themes were related back to the trial progress, and ultimate trial status, as of August 2015.
Human subjects protection
The University of Michigan human subjects review committee deemed this project exempt from Institutional Review Board oversight per 45 CFR 46.101(b). No patient data were collected or used, as the focus of this study was on researchers. Prior to data collection, we provided a written notice informing participants of the research and that their responses were completely voluntary.
Results
Characteristics and overview of the six different clinical trial designs (SHINE, SHINE hemorrhage, ESETT, ICECAP, ARCTIC, and ProSPECT) are as follows.
Table 1 provides a summary of the different clinical trial designs. Because SHINE hemorrhage grew out of SHINE, their results are combined. Each trial had a team composed of PIs, clinical team members, a trial statistical team, and designated adaptive design lead statisticians. The process interactions throughout the trial development process were relatively similar across trials.
Summary of five trials in the ADAPT-IT project.

ADAPT-IT: Adaptive Designs Accelerating Promising Treatments Into Trials; FTF: face-to-face; CTC: concept teleconference.
This table presents a summary description of five adaptive clinical trials (one with a sub-project).
General changes in attitudes over ADAPT-IT process through the longitudinal development
Across all six trials, changes in beliefs about some aspects of ACT changed over the course of the ADAPT-IT project. Table 2 reports the attitudes after the final face-to-face meeting, along with a difference score from baseline. Highlights include the instrument items about beliefs regarding whether NIH review panels, the FDA, and clinicians will understand the ACT design as valid. Results from these items revealed no change over time that we deemed as meaningful. Ratings of clinicians’ understanding of the design were lowest with a mean of 50.4. Significant changes include beliefs about acceptance of ACT designs as valid by NIH review panels (mean difference = 3.6, 95% CI = 0.4 to 6.7) and clinicians (mean difference = 4.0, 95% CI = 0.5 to 7.4). Attitudes about the ACT design meeting the scientific (mean difference = 4.2, 95% CI = 1.7 to 6.8) and medical goals (mean difference = 5.8, 95% CI = 2.9 to 8.6) of the studies also improved. As expected, attitudes about traditional designs meeting scientific and medical study goals did not change. In addition, the results demonstrated a favorable change of attitudes about ethical disadvantages of ACTs from both the patients’ (mean difference = 5.3, 95% CI = –8.4 to –2.2) and society’s perspectives (mean difference = 3.7, 95% CI = –7.2 to –0.3). Conversely, changes in attitudes about ethical advantages from these two perspectives were not significant and were not meaningful. Attitudes from the perspective of the researcher did not differ significantly from baseline.
Change over time in attitudes about adaptive clinical trials, mean VAS scores for all trials (n = 53).

VAS: visual analog scale; FTF: face-to-face; SE: standard error; CI: confidence interval; NIH: National Institutes of Health; FDA: US Food and Drug Administration.
The response scale for all items was a visual analog scale with a range from 0 to 100. The table compares the baseline and final VAS score for each item on the survey. Please see Online Appendix for scale. Results are aggregated across all trials.
Next, we report on each trial individually, summarizing the design development process for each, observations by the process evaluation team, and attitudes about adaptive designs, as assessed through the VAS at baseline and after the final face-to-face meetings. The complete VAS means for each trial appear in Table 3.
Change in attitudes and final impression within each trial development process.

VAS: visual analog scale; FTF: face-to-face; NIH: National Institutes of Health; FDA: US Food and Drug Administration.
The table presents the mean VAS for each item at final face-to-face meeting along with the change from baseline. Results appear for each of the five trials. The SHINE hemorrhage sub-project is included in the SHINE data. For example, for the ESETT trial, participants rated a traditional design with a 48 (consistent with possibly), and this represents a large 29 point drop from the initial meeting where they indicated a 77 that was consistent with the traditional design being likely to meet the scientific goals. The full scale of VAS used is available with the instrument in the Online Appendix.
Stroke Hyperglycemia Insulin Network (SHINE): qualitative findings of the development process
The definitive design of SHINE has previously been described. 12 The first face-to-face meeting of the SHINE trial included 18 individuals, comprised NETT clinical and statistical leaders, adaptive design consultants, clinical team members, NIH partners, a patient advocate, and mixed methods process evaluators. Key discussion points for the meeting were trial requirements and goals and adaptive design principles and preliminary ideas. The meeting involved an extensive discussion of efficiencies gained with adaptive trials in addition to barriers and challenges. Analysis of observational field notes of the meeting identified several themes, including participants feeling rushed and not having clear expectations. Small group sessions were generally productive, except when adaptive design experts huddled together. During those times, other participants were not engaged in the process. In addition, tension arose relative to the statistical assumptions between consultant and academic statisticians. Based on the process evaluation, key recommendations included protecting time to review the agenda, allowing general discussion, and discussing next steps.
A subsequent concept teleconference included discussion of the original trial design, proposed adaptive design, and hierarchical modeling. Concerns arose over logistics of the trial. Three action items arose from the meeting: (1) demonstrate the effect of multiple O’Brien Fleming interim analyses, (2) examine more aggressive futility boundaries, and (3) discuss hierarchical modeling in a smaller group. To move forward, a smaller executive workgroup was formed to develop the adaptive design. The final step was a second face-to-face meeting to review the near-final plan for SHINE, discuss adaptive design efficiencies and challenges, and determine next steps. Overall, the meeting was more collegial and engaged individuals with differing viewpoints. Real-time summaries of the conversation appeared helpful, as the team followed the agenda more closely than during previous meetings.
SHINE VAS attitudinal changes
Through the design development process, stakeholder changes in attitudes about adaptive trials were statistically significant for two VAS items: (1) whether an NIH grant review panel will understand the adaptive design and (2) whether an adaptive design will increase the overall efficiency of the research. Both of those items reflected a change towards less favorable views of adaptive designs. Other items concerning external stakeholder understanding, trial design, and ethical aspects were unchanged. Findings from the process evaluation of SHINE indicated the importance of working on the adaptive trial design at a very early stage. As is, SHINE appeared too well developed before beginning the ADAPT-IT design process, which may have in part been related to an imminent funding decision by NINDS. Developing and clarifying the relationship between trial and consultant statisticians may also have enhanced the design development process. In addition, it became evident that the consultant and academic biostatisticians needed to have highly technical discussions about modeling, model assumptions, and simulation. Initially, these discussions occurred with the clinician-researchers in the room; subsequently, the group set aside time for these discussions to occur between the statisticians, and they were optional for motivated clinicians.
SHINE lessons learned
The initial ADAPT-IT charge was to build a concurrent phase-II trial (also known as SHINE hemorrhage) in subarachnoid hemorrhage (SAH) and intracranial hemorrhage (ICH) that would borrow strength from the ongoing phase-III SHINE trial of ischemic stroke. The main SHINE trial had just been approved for funding prior to the first face-to-face meeting and as such, several elements of the statistical plan had yet to be finalized. As this was one of the first ADAPT-IT meetings, the focus shifted from SHINE hemorrhage to developing a more efficient, alternate interim analysis plan, using Bayesian predictions to set thresholds for futility and efficacy. This was developed and published, but not implemented (known as Shadow SHINE). 13 The NETT team agreed to preserve the actual SHINE database in such a way that the Shadow SHINE algorithm could be applied to the actual trial data observed in SHINE, and we could observe at what time points different decisions would be made regarding continuing the trial or terminating for efficacy or futility. One key distinction between the Shadow SHINE trial, which represented a major difference in philosophy, was that the trial would terminate if it was extremely unlikely that the intensive insulin group was superior. In the actual SHINE trial, if the control treatment (usual care of glucose) is trending towards potential superiority, the two-sided final hypothesis allows for the trial to continue and to definitively prove that intensive insulin management is inferior. The need to have a definitive answer either way was identified by the SHINE study team as a major priority after the two designs were formally compared after ADAPT-IT, but this was not discussed at length during the actual ADAPT-IT meetings. This illustrated a key lesson regarding the early involvement and collaboration between the network biostatisticians (who usually conducted trials with two-sided hypothesis tests for a variety of reasons), and the consultant biostatisticians (who usually designed trials with one-sided hypothesis tests). The SHINE hemorrhage design was simulated and the code is available for the R statistical environment (http://hdl.handle.net/2027.42/134516).
Acute Rapid Cooling Therapy for Injuries of the Spinal Cord (ARCTIC): qualitative findings from the development process
The concept for the ARCTIC trial has previously been described. 14 The initial ARCTIC face-to-face meeting included 28 individuals in a similar make-up to SHINE. The discussion also focused on trial requirements, goals, adaptive designs principles, and efficiencies. A key insight of the observation was that participants did not enter with a clear expectation of what the meeting would accomplish. Despite a pre-specified presentation format and agenda, the meeting drifted from the agenda as the leaders encountered a number of tangential discussions about less important topics that were unproductive. Similar to the SHINE trial, compared to the design experts, others were less engaged in discussions and developed misconceptions of the design planning. Based on observations from the ARTIC face-to-face meeting, the process evaluation team recommended holding adaptive design discussion in an open forum for all to observe and gain understanding. Additional process recommendations were to have a dedicated moderator to attend to the agenda, to allocate discussion time following presentation sections, and to consider small group time to better engage all participants.
The concept teleconference discussion for ARCTIC was similar to SHINE. The meeting leaders, however, explained the purpose of the meeting and repeatedly encouraged questions. A scientific discussion ensued around responsive adaptive randomization over the duration of cooling and changing the time. Refinement of the design continued in the second face-to-face meeting. A main point of discussion was safety outcomes and enrichment plans if the initial 0 to 6-h enrollment time did not show effectiveness in any arms. By this time, participants were more familiar and open with each other. Despite some disagreement between consultant statistician and academic biostatistician teams, they were confident that they could resolve the details in a way that was mutually satisfactory. The consultant and academic biostatistician teams held a subsequent call to discuss the details of the design and simulations.
ARCTIC VAS attitudinal changes
ARCTIC VAS results showed a significantly improved perception of whether the FDA will understand the adaptive design, relative to regulatory approval. Attitudes also improved significantly about whether designs would meet scientific and medical goals of the study for both traditional and adaptive designs. Changes in other items and attitudes concerning ethical aspects were not statistically significant. Regarding the ARCTIC process evaluation results, a lesson learned through the design development process was that decisions about grant submission and timing needed to be more clearly communicated to the team, particularly before doing additional simulation work.
ARCTIC lessons learned
This trial had been submitted once before ADAPT-IT. The initial post-ADAPT-IT submission received mixed peer review. As a II/III design, it was submitted under a confirmatory trials mechanism. As such, the review panel did not feel that therapeutic hypothermia had sufficient preliminary pre-clinical and clinical evidence to justify a “confirmatory” clinical trial. The design did have futility stopping built in, such that it would most likely terminate if none of the durations emerged to be superior to a non-cooling arm. On the second submission, phases II and III were pulled apart from a funding perspective; the ability to quantitatively pick a duration and decide to progress to phase III remained. This time, only the exploratory phase trial was submitted under that mechanism. Again, the design itself was reviewed favorably, but the therapeutic hypothermia was criticized. At this point, plans for an additional submission of this design are uncertain, but the machinery of the design could be applied to other applications.
ESETT: qualitative findings from the development process
The ESETT concept and design have been previously described.15–17 The initial ESETT face-to-face meeting consisted of 24 individuals. Discussion points were similar to the ARTIC trial but added issues of dosage and rules for dropping and inferior drug or dose. It further introduced a discussion of exception from informed consent because enrolled patients would be in status epilepticus. An issue observed during this meeting was the need to better define the clinical question in the study. Evaluators recommended a small workgroup to address the issue and resolve ambiguity. Tension arose concerning statistical assumptions during this meeting between statisticians and physicians, particularly concerning the statistical expertise of physicians. The subsequent concept teleconference resolved design features, such as adaptive randomization and dropping underperforming arms noted in an interim analysis. A strong moderator kept the conversation focused by explaining the purpose and encouraging questions repeatedly. The second face-to-face meeting reviewed the proposed Bayesian design. A strong collaboration between the consultant and academic statisticians prior to the meeting resulted in a more refined design with ownership by all statistical parties. The value of encouraging this planning collaboration was a major lesson learned. In particular, focusing early on design led to a better process and grant.
ESETT VAS attitudinal changes
The results from the ESETT VAS were consistent with the observation about focusing early on the design and indicated a relatively positive view of adaptive trials from baseline to final face-to-face meeting. Only one item changed significantly from baseline—the stakeholders reported more improved attitudes about whether the adaptive design will meet scientific goals of the study. Results concerning stakeholder understanding, other trial design aspects, and ethical concerns were positive and unchanged throughout the design development.
ESETT lessons learned
Similarly, the process for developing ESETT was generally non-controversial from the early ADAPT-IT meetings on. Given that patients with status epilepticus needed to be treated with something, and three different agents were currently used, a trial using response-adaptive randomization (RAR) to allocate patients to the better performing treatments tended to improve the study’s ability to get a useful answer and more effectively treat the patients within the trial. ESETT was submitted for funding and approved on the first submission to NINDS. It is currently enrolling patients within the design that was initially conceptualized in the first face-to-face meeting, although the details of the logistics and exact performance of the model were worked out over time.
ICECAP: qualitative findings from the development process
The ICECAP face-to-face consisted of five FDA partners and 26 individuals in similar roles as other meetings. The discussion added points about the proposed design, which included three treatment durations, hierarchical modeling, and adaptive randomization. The process evolved in that the PI, a senior investigator, served as a strong moderator to focus the meeting and call upon individuals to ensure everyone’s input was heard. Additional changes included real-time summaries by a scribe, structured breaks, and discussion time. The changes to the meeting process seemed productive and effective. Participants appeared to highly value input from FDA participants. Based on the process evaluation, the major recommendation was to evaluate continuing participation of statisticians.
ICECAP VAS attitudinal changes
Based on VAS results, attitudes about adaptive designs did not change significantly from baseline for the ICECAP respondents. Overall, perceptions of stakeholder understanding, trial design goals, and ethical aspects were positive. Though this adaptive design was extremely complicated, similar to ESETT, it was generally agreed upon as the best way to address the questions of how long to cool cardiac arrest victims and whether cooling was effective at all. Two NIH institutes, NINDS and NHLBI (National Heart, Lung, and Blood Institute), were interested in this project. Several meetings occurred to reconcile the budget, scale, and scope of the project. Both institutes require high-level approval for the submission of grants over US $500,000 per year in direct costs. Currently, work continues with the institutes to allow for the grant submission. Concurrently, the investigators worked with the FDA to further refine and clarify the design within the context of pre-IDE meetings. A full IDE was submitted on 1 April 2016. A new emergency care network focused on neurological disease, cardiac/pulmonary/blood emergencies, and trauma has been announced by NINDS and represents a potential platform for the ICECAP trial.
ProSPECT trial: qualitative findings from the development process
The ProSPECT trial was developed to evaluate the neuroprotective agent, progesterone, in patients with ischemic stroke. With no academic statisticians present and only 1 NIH partner, the initial ProSPECT face-to-face meeting was smaller with 21 individuals. The discussion began with a summary of the ADAPT-IT project to date, including goals, methods, and accomplishments. Relative to other meetings, less time was spent on general principles of ACTs and more time was devoted to the preliminary work and clinical design of the trial. The observations revealed a different atmosphere, which was friendly and interactive. The lead presenter was particularly engaging and light-hearted. A notable difference was the involvement of a pre-clinical scientist, who contributed to issues about translating pre-clinical results and processes to the trial. No issues or controversies arose. The concept teleconference focused on a presentation of the initial ACT design to include a first-stage learning, followed by a confirmatory (pivotal) stage. The team held considerable discussions about simulations and created plans for the first set of simulations. The second face-to-face meeting included the academic statisticians and provided an update of the trial design. A new presentation for this meeting, “Where we are and how did we get here?” summarized the trial evolution, all considerations, and prior suggestions. It promoted collaboration, and the evaluators recommended the presentation for all future final meetings.
ProSPECT VAS attitudinal changes
The VAS baseline and final beliefs about ACTs clearly indicated a preference for adaptive designs initially. Although some items reflected a change that preferred traditional designs from baseline to final face-to-face meeting, none of the differences were statistically significant. The number of responses, however, was small (n = 3). Based on the process evaluation of the ProSPECT design development, a lesson was to brief patient advocates prior to the meeting to enhance their contribution.
ProSPECT represented a different planning process, as the focus was on an exploratory (not confirmatory) trial. This was one of the most complicated designs, and it involved picking both a dose and administration duration of progesterone for stroke. While this agent had an excellent evidentiary basis from a wealth of pre-clinical data for ischemic stroke, an ongoing phase-III NETT trial evaluating progesterone in traumatic brain injury was stopped due to futility. 18 As such, the design was not pursued further. However, it was developed sufficiently, such that it could be used for human or animal exploratory trials of evaluating two linear regimen factors (in this case dose and duration of the therapy).
Summary of attitudes about adaptive designs
Table 4 graphically represents the mean change in attitudes from baseline for each trial. Six of the items in the table were reverse-coded for analysis so that a larger value represents a more favorable attitude towards ACTs. Final attitudes were most favorable towards adaptive trials for ESETT and ProSPECT, and least favorable for SHINE. ESETT, which was funded, experienced the largest change in attitudes over time on average. Changes reflected more positive views about adaptive designs. Change was strongest for the ESETT trial, relative to others when asked about whether the design will meet the scientific goal of the study. Responses reflected less favorable attitudes about traditional trials and a corresponding increase in favorable attitudes about adaptive trials. Another notable difference was evident in the ProSPECT trial, for which an adaptive design was not pursued. Despite strong overall mean final attitudes towards adaptive trials, the patterns of change over time among ProSPECT participants differed from other trials and favored traditional designs. In particular, respondents’ changes in attitudes reflected concerns about adaptive trials regarding the ability of NIH grant reviewers to understand adaptive trials and whether traditional designs will meet the scientific study goals. Although the attitudes shifted towards less favorable views of adaptive designs for ProSPECT, the changes were not statistically significant.
Mean difference in attitudes from baseline for five trials.



VAS: visual analog scale; NIH: National Institutes of Health; FDA: US Food and Drug Administration.
This radar graph plots the mean change from baseline for each of the 21 items on the VAS scale. The area inside the circle marked with zero indicates negative changes for that domain, and the area outside that circle indicates improvements in attitude. For example, improved attitudes are observed from domain 6 through 20 for the ESETT trial. Results are presented for each of the trials to allow for comparison. SHINE hemorrhage is included in the SHINE data.
Stakeholder attitudes also changed over time (see Table 5 for a comparison of stakeholder types). As noted previously, six items were reverse-coded, so a larger number represents more favorable beliefs about ACTs. Across all items, clinicians had the biggest change in attitudes about ACTs. Attitudes improved significantly concerning ACT designs, meeting the scientific (95% CI = 0.5 to 8.3) and medical (95% CI = 1.9 to 24.2) goals of the study. Other items with significant differences favoring ACTs related to ethical disadvantages from the patient’s perspective (95% CI = –26.6 to –3.7) and researcher’s perspective (95% CI = –22.5 to –0.2). Consultant statisticians had the most favorable views of ACTs at baseline and final and tempered some views on ACTs. However, the changes were not statistically significant. On the other hand, academic biostatisticians showed change from baseline to final, but the change generally reflected a less favorable attitude towards ACTs. Significant changes in attitude included items related to the NIH review panel understanding the ACT (95% CI = –24.6 to –5.9), whether the traditional design would meet medical goals (95% CI = 0.5 to 29.1) and produce valid scientific results (95% CI = 1.1 to 26.9), and the efficiency of ACTs (95% CI = –33.7 to –4.1). Finally, other stakeholders (e.g. regulatory officials) developed more positive views of ACTs on average, primarily for aspects related to understanding and accepting adaptive trials, but changes were not significant.
Mean difference in attitudes from baseline for four types of stakeholders.
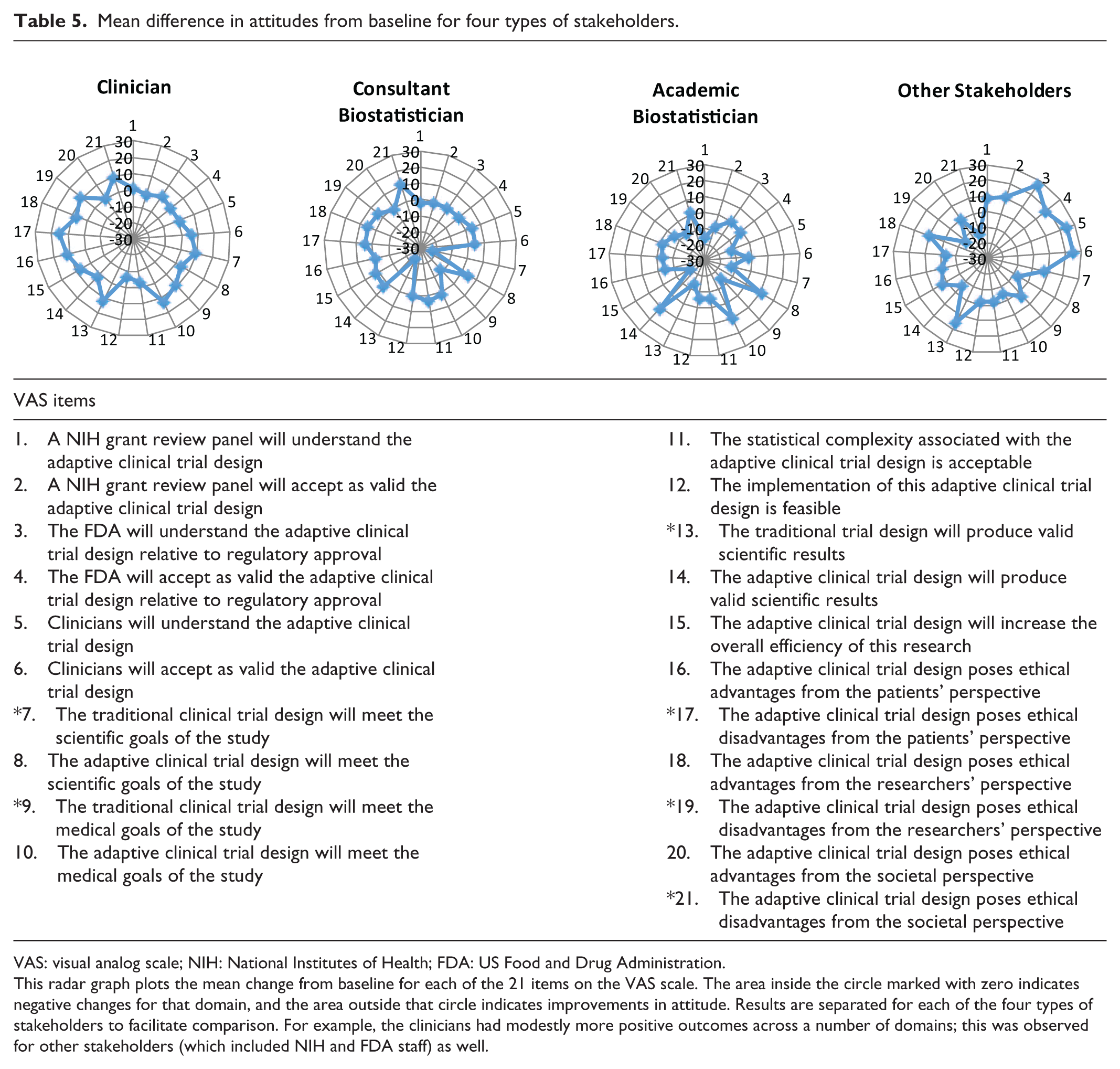

VAS: visual analog scale; NIH: National Institutes of Health; FDA: US Food and Drug Administration.
This radar graph plots the mean change from baseline for each of the 21 items on the VAS scale. The area inside the circle marked with zero indicates negative changes for that domain, and the area outside that circle indicates improvements in attitude. Results are separated for each of the four types of stakeholders to facilitate comparison. For example, the clinicians had modestly more positive outcomes across a number of domains; this was observed for other stakeholders (which included NIH and FDA staff) as well.
Discussion
Within ADAPT-IT, the team collaboratively and longitudinally developed six flexible ACT designs. The development process was similar for all designs in that each followed relatively similar steps. The process included an initial face-to-face meeting, a concept teleconference, smaller working groups, and a second face-to-face meeting. However, the content tended to vary considerably in terms of the initial discussion of adaptive principles, the contention that arose, and the number of working groups. Furthermore, the longitudinal evaluation of the process proved quite valuable in identifying lessons learned and new strategies that were incorporated into subsequent trials and discussions. Recommendations from the iterative evaluation influenced the process. For instance, through the six trials, the conversation strategy evolved to have the PI serve as a strong moderator for meetings and focus more on the clinical design of the trials. The current status of the trials range from well-described concepts (SHINE hemorrhage) to an actual implemented and ongoing trial (ESETT).
Consensus about design
Consensus regarding the ideal design concept was generally elusive. Proponents of more traditional designs and proponents of more flexible adaptive designs generally retained their opinions over the course of the process. However, in certain cases (e.g. ESETT), the flexible adaptive design was favored from the beginning, and stakeholders’ attitudes remained strong throughout the process. ESETT was the one case where consensus between all parties on the suitability and practicality of the adaptive design was generally achieved. Initiating open discussion of an adaptive design early on, and allowing sufficient time for discussion, appeared to facilitate the process to achieve a successful and fundable trial. The design development was a process. Despite differences in opinion, a key advantage was having the same core group of individuals working together to design the adaptive trial.
Introduce adaptive designs from conceptualization
Previous work has highlighted various challenges to planning and implementing adaptive designs in both the academic and industry settings.5,9–11 The results of this longitudinal process evaluation across the six trials suggest that introducing adaptive designs from early conceptualization of the trial is more effective than fitting an adaptive design at later stages. The results also demonstrated that attitudes concerning adaptive trials changed throughout the ADAPT-IT design development process. The trial concepts developed within ADAPT-IT occurred within an evolving process of scientific reflection and revision of the design. That process seemed important to allow for multiple perspectives, contention, and discussion about the design. The key early scientific input of clinicians, biostatisticians, adaptive design experts, and basic scientists generally contributed towards progress.
The ADAPT-IT design development process consisted of an initial face-to-face meeting with all stakeholders, a concept teleconference, a series of working groups, and a final face-to-face meeting. Of the six trials, the original SHINE trial, which was in final development through ADAPT-IT, is ongoing. ESETT was funded on initial submission, using the design conceptualized at the initial face-to-face meeting. It continues enrolling patients. Although consensus about designs largely did not happen, it was facilitated by early introduction of the ACTs at the conceptualization stage.
Limitations
It is necessary to discuss some limitations of this study. First, the ADAPT-IT process assumed that adaptive designs offer a scientific benefit. Nevertheless, the diversity of views about adaptive trials was evident in our analysis. Second, the relatively small sample size of stakeholders participating in the six trial concepts and focus on neurological emergencies does not permit generalizability to the vast number of clinical trials conducted yearly. The conclusions offer guidance to others who may be developing adaptive trials drawn from experiences with these six trial concepts.
Conclusion
This study raises several implications for planning of future trials and consideration of adaptive components. First, though tempting, it is critical to not ignore the development process. Basic tactics, such as having a strong leader to keep the discussion focused and serve as a moderator, appear critical to move the design forward. Other key insights related to the importance of involving all stakeholders during discussions. For example, statistical discussions involving only consultant statisticians with expertise in adaptive designs led to tension between academic statisticians and were counterproductive. Involving patient advocates also required a different approach. Briefing patient advocates about the nature of the trial before meeting can help them to participate in a more meaningful way. Finally, based on the process across all six trials, introducing the adaptive design early was most productive, enhanced collaboration, and ultimately led to a more refined design.
Footnotes
Acknowledgements
The authors gratefully acknowledge Lilly Pritula for her editorial assistance with preparing the manuscript.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Berry reported that he is a part owner of Berry Consultants LLC, a statistical consulting firm that specializes in the design, implementation, and analysis of Bayesian adaptive clinical trials for pharmaceutical manufacturers, medical device companies, and academic institutions; Dr Lewis reported that he is the senior medical scientist of Berry Consultants LLC; both Dr Lewis and the Los Angeles Biomedical Research Institute are compensated for his time.
Ethical approval
The University of Michigan human subjects review committee deemed this project exempt from Institutional Review Board oversight per 45 CFR 46.101(b).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The ADAPT-IT project was supported jointly by the NIH Common Fund and the FDA, with funding administered by the National Institutes of Neurological Disorders and Stroke (NINDS; U01NS073476). The NETT Network Clinical Coordinating Center (U01NS056975) is funded by the NINDS. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Informed consent
No patient data were collected or used as the focus of this study was on researchers. The authors provided a written notice prior to data collection notifying participants of the research and their responses were completely voluntary.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.