
Abstract
Uveal melanoma (UM) is the most common intraocular malignancy in adults. So far, no systemic therapy or standard treatment exists to reduce the risk of metastasis and improve overall survival of patients. With the increased knowledge regarding the molecular pathways that underlie the oncogenesis of UM, it is expected that novel therapeutic approaches will be available to conquer this disease. This review provides a summary of the current knowledge of, and progress made in understanding, the pathogenesis, genetic mutations, epigenetics, and immunology of UM. With the advent of the omics era, multi-dimensional big data are publicly available, providing an innovation platform to develop effective targeted and personalized therapeutics for UM patients. Indeed, recently, a great number of therapies have been reported specifically for UM caused by oncogenic mutations, as well as other etiologies. In this review, special attention is directed to advancements in targeted therapies. In particular, we discuss the possibilities of targeting: GNAQ/GNA11, PLCβ, and CYSLTR2 mutants; regulators of G-protein signaling; the secondary messenger adenosine diphosphate (ADP)-ribosylation factor 6 (ARF6); downstream pathways, such as those involving mitogen-activated protein kinase/MEK/extracellular signal-related kinase, protein kinase C (PKC), phosphoinositide 3-kinase/Akt/mammalian target of rapamycin (mTOR), Trio/Rho/Rac/Yes-associated protein, and inactivated BAP1; and immune-checkpoint proteins cytotoxic T-lymphocyte antigen 4 and programmed cell-death protein 1/programmed cell-death ligand 1. Furthermore, we conducted a survey of completed and ongoing clinical trials applying targeted and immune therapies for UM. Although drug combination therapy based on the signaling pathways involved in UM has made great progress, targeted therapy is still an unmet medical need.
General features
As the most common intraocular malignancy, the annual incidence rate of uveal melanoma (UM) is 4.3 cases per million population. 1 However, the incidence rate varies among countries owing to differences in diagnosis and classification criteria, as well as risk factors associated with UM, like fair skin, light iris color, inability to tan etc. 1 The incidence rate of UM in the United States is around 5.1 cases per million population per year. 2 In Europe, the incidence rate increases from southern to northern Europe, with two cases per million in Spain and southern Italy to up to eight cases per million in Norway and Denmark, and an annual range of 1.3–8.6 cases per million. 3 The incidence rates of UM in Australia and New Zealand are as high as those in the United States and European countries, at 9.8 and 9 cases per million people per year, respectively.4,5 The incidence rate is relatively low in Asia, including South Korea (0.42 cases per million population per year 6 ) and Japan (0.64 cases per million population per year 7 ), and in Africa (0.3 cases per million population per year 8 ). Incidence increases noticeably up to the age of 55 years, and levels off after the age of 75 years. 3 It is also higher in males, with incidence rate ratio of 1.22 compared with that in females. 3 Risk factors associated with UM include age (50–70 years), fair skin, light iris color (blue or gray), and sensitivity to sunburn.9,10
UM arises from melanocytes in the uvea, 90% of which involve the choroid, and 6% of which are confined to the ciliary body and 4% to the iris. 11 The most common clinical symptoms of UM include blurred vision, visual field loss, photopsia, or change in iris color; about 30% of UM are asymptomatic and detected on routine examination. 12 Primary intraocular tumors can be detected using fundoscopy and MRI imaging (Figure 1), and are treated effectively using radiation plaque therapy, which achieves tumor control in 98% of eyes, with 95% globe salvage. 13 Enucleation is indicated for advanced UMs, with optic nerve involvement or orbital invasion. 14 However, approximately half of the cases will develop metastasis, predominantly (90%) to the liver.15,16 The 10-year metastasis rate varies among UM patients depending on the tissue of origin; it is 33% for ciliary body melanoma, 25% for choroidal melanoma, and 7% for iris melanoma.17,18 Once metastasis occurs, the median progression-free survival (PFS) and overall survival (OS) is 3.3 months and 10.2 months, respectively. 19 Another systematic review demonstrated that the median OS across all treatments for metastatic UM is 12.8 months. 20
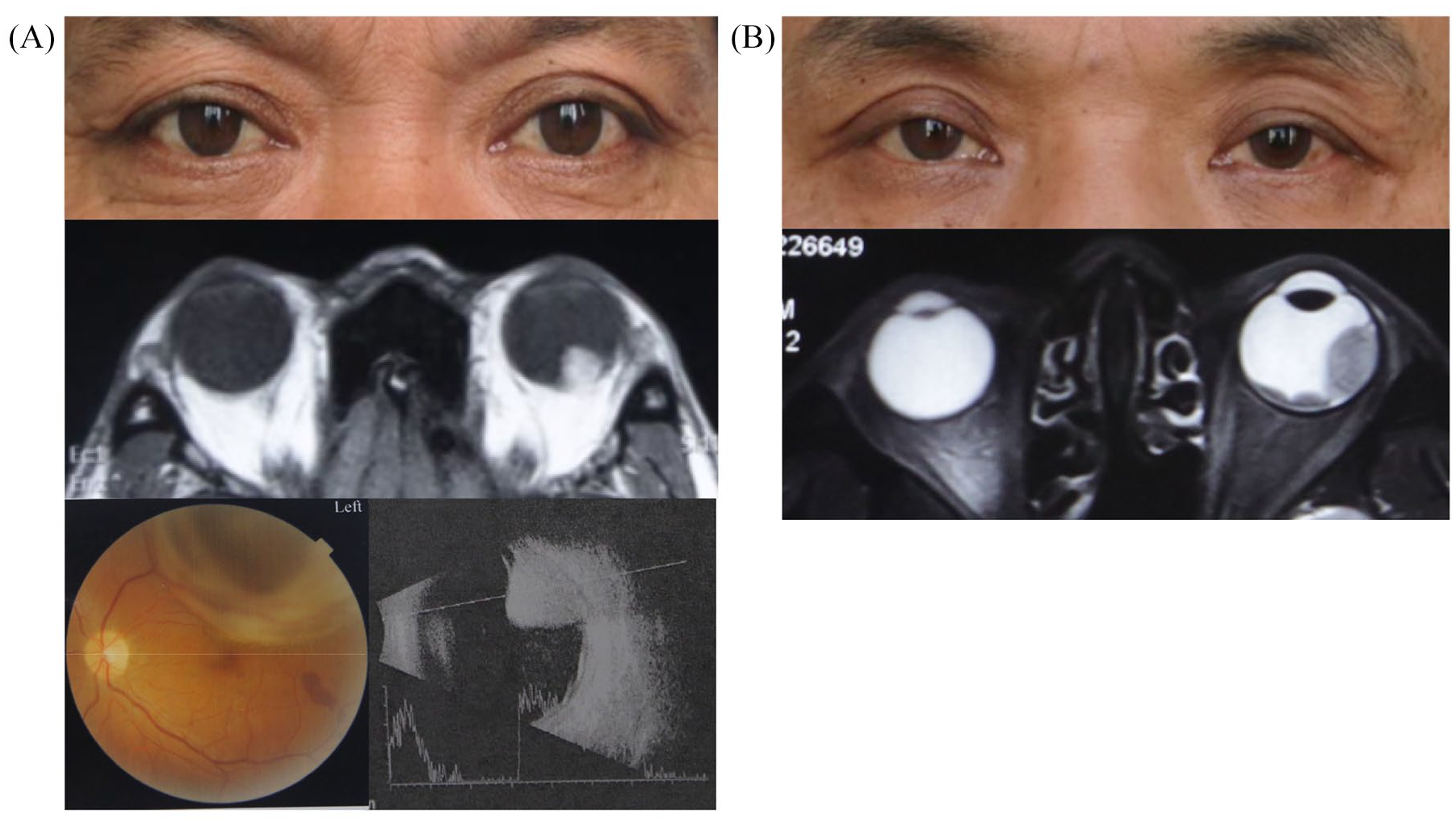
Clinical manifestation and imaging examinations of typical UM patients.
UM is often initiated by a GNAQ or GNA11 mutation with low tumor mutational burden, unlike cutaneous melanoma, which is usually triggered by a BRAF or NRAS mutation with multiple single-nucleotide polymorphisms. 21 The classification of UM has been updated recently into four molecularly distinct subtypes: (a) poor-prognosis monosomy 3 (M3) with BRCA1-associated protein-1 (BAP1) aberration; (b) M3 without BAP1 aberration; (c) better-prognosis disomy 3 (D3) with serine/arginine-rich splicing factor 2 (SRSF2)/splicing factor 3B subunit 1 (SF3B1) mutation; and (d) D3 with eukaryotic translation initiation factor 1A, X-linked (EIF1AX) mutation. 22 BAP1 loss correlates with a global deoxyribonucleic acid (DNA) methylation status, dividing M3-UM into subsets with different genomic aberrations, transcriptional patterns, and clinical outcomes. 22 Regarding D3-UM, SRSF2/SF3B1-, and EIF1AX- mutants, tumors have distinct DNA methylation profiles and somatic copy-number alterations, representing low- and intermediate-risk mutation subtypes, respectively. 23 Based on new discoveries on the pathogenesis of UM, novel therapeutic options are emerging, but no effective standard treatment is available for advanced and metastatic UM patients.
Molecular pathogenesis
Gene mutations
Dysregulation of G-protein signaling
Guanine-nucleotide-binding proteins (G proteins) are a class of hydrolases that act as molecular switches to transduce signals from extracellular stimuli perceived by G-protein-coupled receptors (GPCRs) to the cell interior.24–26 G proteins are grouped into two categories: monomeric small GTPases (e.g. Ras), and heterotrimeric G protein complexes, formed by Gα, Gβ, and Gγ subunits. 27 The Gα subunit can bind to either guanosine triphosphate (GTP) or guanosine diphosphate (GDP). The GDP-bound state connects with β and γ subunits to form a trimeric complex. When GDP is exchanged with GTP, initiated by guanine-nucleotide exchange factors (GEFs), the Gα subunit is activated, dissociates from the receptor and Gβγ subunits, and triggers downstream signaling cascades. The GTPase activity of the Gα subunit hydrolyzes GTP to GDP, and signal transduction is terminated 28 (Figure 2). This inactivation process is catalyzed by regulators of G-protein signaling (RGSs), such as GTPase-accelerating proteins (GAPs). 29 GPCRs and the downstream G-protein signaling are both important targets for current drug discovery, since GPCRs strongly impact a wide range of physiological and pathological conditions.30,31 Pharmacological targeting of GPCRs has become increasingly attractive as the detailed molecular machinery of GPCRs in tumor development is being elucidated. Indeed, Degarelix®, which is a gonadotropin-releasing hormone (GnRH) receptor antagonist, has been approved for patients with advanced prostate cancer. 32 Vismodegib and sonidegib, which are smoothened (SMO) receptor inhibitors, are approved for the treatment of basal cell carcinoma. 32

Schematic representation of G-protein movement and the Gα subunit inhibition by YM‑254890 and FR900359.
There are 20 different kinds of G-protein α-subunits, divided into four families Gαs, Gαi, Gαq, as well as Gα11, Gα12, and Gα13. Each family exerts specific functions on the regulation of certain sets of downstream targets. 33 Interestingly, most UM cases have mutually exclusive mutations in guanine-nucleotide-binding protein G(q) subunit alpha (GNAQ) and guanine-nucleotide-binding protein G(11) subunit alpha (GNA11).34,35 GNAQ encodes Gαq and GNA11 encodes Gα11. Gαq/11 plays an important role in regulating cellular functions and pathological processes of diseases, such as insulin-stimulated glucose transport, platelet aggregation, heart failure, and cancer.36–39 It has been reported to couple with certain GPCRs, such as the endothelin 1 receptor, angiotensin 2 receptor type I, α-1 adrenergic receptors, and vasopressin type 1A and 1B receptors. 40 GNAQ or GNA11 mutations occur in nearly 80~93% of UM patients, each at a proportion of 20~50% and 43~60%.41–44 The most predominant mutation occurs in exon 5 (Q209; >70%), which entirely cripples the intrinsic GTPase activity of the Gα subunits, resulting in a persistent active state. 45 Mutations in exon 4 (R183) have been detected in a small fraction of cases (<10%), reported as 40% GTP bound, indicating that it maintains some GTP hydrolysis activity. 35 Constitutively activated mutants Gαq and Gα11 drive abnormal proliferative signaling via the extracellular signal-intracellular signaling pathway, similar to the activating mutations of BRAF observed in cutaneous melanoma. 46 In particular, these mutations could not predict the outcome, survival, or risk of metastasis in UM, which indicates oncogenic driver mutations in Gαq and Gα11. 47 However, the GNA11 Q209 mutation is more frequently observed in metastatic UM (57%) and tumors involving the ciliary body, or with mutations in BAP1, suggesting that compared with GNAQ, mutation in GNA11 is correlated with a higher risk of metastasis.43,44 Benign blue nevi can also harbor GNAQ and GNA11 mutations (83% and 7% respectively), which indicates that G-protein mutations are an early event in UM tumorigenesis.34,35,48
Apart from mutations in G proteins, variations of the GPCR itself and its effectors can also alter G-protein signaling and are related to UM pathogenesis. The cysteinyl leukotriene receptor 2 (CYSLTR2) gene, which encodes a GPCR that activates Gαq, has been found to have a substitution (p.Leu129Gln) in 3% of UM samples. 49 Whether CysLT2R binds to Gα11 is unknown; however, the p.Leu129Gln mutation mediates activation of signaling pathways that are convergent with those activated by GNAQ and GNA11 oncogenic mutations. 49 This hotspot mutation drives aberrant cell growth in vitro and promotes tumorigenesis in vivo. 49 Apart from UM, the same hotspot mutation in CYSLTR2 has also been identified in blue nevi. 50 A whole-genome sequencing study on 28 UM tumors or primary cell lines revealed the presence of the mutation p.D630Y in phospholipase-C beta 4 (PLCB4) in two samples (4%). 51 In addition, 4% of UM samples have the p.K898N mutation in phospholipase-C beta 3 (PLCB3), which is localized in the C-terminal domain linker and plays a vital role in GNAQ activation. 51 PLCB4 and PLCB3 are both downstream effectors of GNAQ/GNA11. 51 Notably, all the CYSLTR2, PLCB4, and PLCB3 mutations exclusively exist with GNAQ and GNA11 mutation, occur within the same pathway, and provide the possibility of using novel drugs to target different mutant forms.
As G-protein signaling plays a crucial role in UM, it is important to understand the downstream pathways of Gαq and Gα11 in order to develop effective treatments for UM patients. The downstream effectors include phospholipase-C beta (PLCβ), PKC, Rho/Ras-related C3 botulinum toxin substrate 1 (Rac1), ARF6, phosphoinositide 3-kinase (PI3K), and β-catenin.
The best-known downstream signaling cascade initiated by Gαq/11 involves the activation of PLCβ and the consequent increase in levels of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). 52 IP3 leads to a rapid increase in cytoplasmic Ca2+ levels, hence modifying a series of calcium-regulated events. DAG stimulates the phosphorylation of PKC and guanyl-releasing protein 3 (RasGRP3) at the plasma membrane. 53 Phosphorylated PKC and RasGRP3 activate RAF/MEK/ extracellular signal-related kinase (ERK), a type of mitogen-activated protein kinase (MAPK) cascade, to regulate several cellular processes, including differentiation, proliferation, survival and apoptosis. 54 The ERK1/2 pathway is reported to be upregulated in 45–86% of primary UM tumors.55,56 It is worth noting that continuous activation of the MAPK cascade may not require mutant Gαq/11, since silencing of GNAQ expression did not suppress ERK activity in GNAQ-mutant UM cells. The activation of the MAPK cascade in UM can be caused by secondary genetic alterations during disease progression. 57
Although PLCβ is considered the canonical downstream effector of Gαq/11, additional effectors of Gαq/11 have been discovered. Particularly relevant to UM, Gαq stimulates RhoA and Rac1 small GTPase-induced signaling via binding to p63RhoGEF and Trio, members of the large Rho guanine-nucleotide exchange factor family.58,59 The signaling of the Rho family regulates cytoskeleton-dependent processes and transitions for a particular type of invasiveness during cell migration. 60 Pathways downstream of RhoA/Rac1 are likely to deliver the mitogenic signals from Trio in the cytoplasm and then to the nucleus. Trio activates other signaling nodes of MAPKs, JNK, and p38, to influence the transcription factor activator protein 1 (AP1), which controls the expression of several growth-promoting genes. 61 Moreover, Gαq/11 stimulates nuclear translocation of Yes-associated protein (YAP), a critical component of the Hippo signaling pathway, promoting actin polymerization. Gαq/11 mutations promote YAP-associated growth of UM cells.62–64 Recently, it has been reported that YAP is required for tumor lymph node metastasis through the upregulation of genes in the fatty-acid oxidation (FAO)-related signaling pathway. Overexpression of YAP results in cytoskeletal rearrangement and induces tumor migration via regulating F-actin/G-actin turnover.65,66
PI3Ks are regulated by a variety of upstream activators, including GPCRs (Gβγ) and small GTPases from the Ras and Rho families. They catalyze the formation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) from phosphatidylinositol (4,5)-bisphosphate (PIP2), and PIP3 phosphorylates and activates Akt at the plasma membrane to promote cell proliferation and survival. 67 The PI3K/Akt pathway regulates cell growth and survival in UM, and is abnormally activated in more than 50% of patients. 68 It is negatively regulated by phosphatase and tensin homolog (PTEN), which reverses PIP2 conversion to PIP3. Although PTEN mutations are not common in UM, 59% of UM cases harbor PTEN gene deletion or have attenuated gene expression. Importantly, PTEN expression loss is associated with shorter disease-free survival of UM patients. 69
The newly identified small GTPase ARF6 is an important mediator of endocytosis and recycling of membrane receptors such as GPCRs and cadherin–catenin complexes. 70 In UM cells, it is a direct downstream effector of Gαq signaling, thus regulating Gαq and β-catenin trafficking. 71 Oncogenic Gαq redistributes into the cytoplasm and forms a complex with the guanine-nucleotide exchange factor GEP100 and ARF6, which acts as a signaling cytoplasmic vesicle. Inhibition of ARF6 in UM cells reduced cell proliferation, as well as activation of all downstream signaling targets PLCβ, MAPK, Rho, Rac, and YAP. 71 Thus, ARF6 provides a novel potential therapeutic target for UM (Figure 3). The interaction between ARF6 and Gα11 remains unknown.

Gαq/11 signaling pathways.
Based on the dysregulation of G-protein signaling in UM, a few transgenic animal models have been developed. The first model was realized by the expression of GNAQQ209L, manifesting as increased neoplastic proliferation in choroid, dermal nevi, and other melanocytic sites, with 94% lung metastasis. 72 Combining GNAQ/11Q209L transgenesis with mutant Tp53, animals led to development of melanocytic tumors, including UM with near-complete penetrance. 73 Recently, a mouse model with melanocyte-specific GNA11Q209L expression with or without BAP1 loss has been generated. Pigmented neoplasms were developed from melanocytes of the skin, eye, leptomeninges, lymph nodes, and lungs. 74 These animal models are considered excellent tools to study molecular and genetic characteristics in UM.
Chromosome 3 and BAP1
Since GNAQ/GNA11 mutations do not predict patient outcomes, the metastasis in class 2 tumors usually involves additional molecular alterations. Monosomy of chromosome 3 and gains of chromosome arm 8q can generally co-occur 75 and are largely associated with UM metastasis and poor prognosis. Mutations in BAP1, a tumor suppressor gene (TSG) located at chromosome 3p21.1, have been identified in approximately 45–47% of UM lesions and in 84% of metastasizing tumors.41,76,77 Notably, inactivation mutations of BAP1 occurred in the majority of class 2 metastasizing tumors, but not in class 1 tumors. 77 BAP1 mutations have been previously found in a variety of cancers, including breast cancer, lung cancer, malignant pleural mesothelioma, cutaneous melanoma, and meningioma.78–81 In UM, BAP1 mutations are accompanied by loss of one copy of chromosome 3 in somatic cells, which supports the ‘two hit’ model that inactivation of BAP1 is associated with metastasis in UM. 77
BAP1 is a deubiquitylase that forms complexes with a variety of proteins and plays important roles in cellular pathways, including DNA damage response (DDR), cell cycle, cellular differentiation, and cell death. 82 BAP1 protein was initially identified in a yeast two‑hybrid screen for its interaction with the tumor suppressor protein breast cancer type 1 susceptibility protein (BRCA1). 78 BRCA1 forms a heterodimer with the really interesting new gene (RING) domain of BRCA1-associated RING domain 1 (BARD1) protein, and this complex has E3 ubiquitin ligase activity that regulates DDR. 83 BAP1 modulates the E3 ligase activity of this complex via binding and deubiquitylating BARD1, and thus, regulates the DDR process. 84 BAP1 is also involved in cell-cycle control via host-cell factor-1 (HCF-1). 85 In addition, BAP1 directly deubiquitinates and stabilizes Krüppel-like zinc-finger transcription factor 5 (KLF5) to promote cell-cycle progression. 86 Furthermore, BAP1 binds additional-sex-combs-like 1 (ASXL1) protein through its carboxyl terminus to form the polycomb repressive deubiquitylase (PR‑DUB) complex that specifically removes monoubiquitin from histone 2A (H2A). 87 Ubiquitylation of H2A is a key mechanism for the polycomb repressive complex 1 (PRC1) to silence gene expression. 88
UM cells with BAP1 depletion exhibit stem-cell-like characteristics. 89 These include loss of morphological differentiation as revealed by downregulation of microphthalmia-associated transcription factor, dopachrome tautomerase, and tyrosinase, as well as upregulation of genes characterizing stem cells. 89
SF3B1, SRSF2 and EIF1AX mutations
In UM, SF3B1 carrying a heterozygous point mutation, predominantly at p.R625, K666, and K700, has been reported in ~25% of UM patients.90–92 This mutation alters the splicing process of certain messenger ribonucleic acid (mRNA) transcripts, but its tumorigenic role remains unclear.91,92 Heterozygous in-frame deletions in another spliceosome factor, SRSF2, have been detected in 5% of UM cases, affecting amino acid residues 92–100. 93 Recently, mutations in EIF1AX (p.N4S), located in the X chromosome, have been detected in 13% of UM patients. 94 EIF1AX encodes a eukaryotic translation initiation factor 1A (eIF1A) that regulates the translation initiation process. Several elements of this process are known to be misregulated in tumorigenesis.95,96 Mutations in EIF1AX have been found in many cancer types and were associated with worse prognosis when coupled with mutations of the Ras family.97,98 In UM, EIF1AX mutations have been reported to be non-truncating and heterozygous. However, only mutant mRNA transcripts are expressed in UM, indicating that the wildtype copy of EIF1AX might be epigenetically inactivated. 91 These mutations are mutually exclusive of each other and of BAP1 mutations in almost all UM cases. 23 Mutations in SF3B1 and SRSF2 are mainly associated with a late-onset metastatic risk, while EIF1AX mutations are associated with low metastatic risk.91,99
Epigenetic alterations
Epigenetic events such as DNA methylation and histone modification involved in the initiation and progression of UM may silence TSGs or activate oncogenes, including noncoding RNAs. 100 The interplay between epigenetic alterations affects the regulation of transcription and/or translation of many key genes and pathways that contribute to UM.
DNA methylation, one of the key epigenetic mechanisms, is shown to be involved in regulation of several UM-related genes. For example, the Ras association domain family 1 isoform A (RASSF1A) gene, located on chromosome 3p21.3, encodes a protein that plays a significant role in apoptosis, cell-cycle regulation, and microtubule stability.101,102 Methylation of the two CpG islands spanning its promoter inactivates this gene and leads to loss of G1/S phase control.103,104 Downregulation of the RASSF1A protein frequently occurs in UM. 105 Other studies have found that cyclin-dependent kinase inhibitor 2A (p16INK4a) is frequently inactivated by DNA methyltransferase 1 (DNMT1)- and DNMT3b-mediated hypermethylation in both primary UM and cell lines. 106 Thus, the downregulation of p16INK4a could be relieved by the demethylating drug 5-aza-2-deoxycytidine.107,108 Interestingly, metastasis is more common in patients possessing a methylated p16INK4a promoter. 108 The TSG Ras and EF-hand domain-containing protein (RASEF) gene is also hypermethylated in UM. It has been reported that lack of RASEF expression in 35 primary UM samples and 11 UM cell lines was due to the methylated promoter. 109 Hypermethylation of the genes Decoy receptor 1 (DcR1) and DcR2 that encode the tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptors has been detected in UM. 110 Recent studies have shown that DNA methylation is also involved in the regulation of other UM-related genes such as RARB, TIMP3, EFS, PTEN, SYK, TNFSF10D, LOX, and COL1A2.111–116
Histone modification includes histone methylation, acetylation, ubiquitination, phosphorylation, and so on. Methylation of histone could be an oncogenic event. It has been reported that impaired class II transactivator (CIITA) transcript levels are associated with high rate of trimethylated histone H3 lysine 27 (H3K27me3) binding to its promoter. 117 The abnormal binding was realized by the histone methyltransferase enhancer of zeste homolog 2 (EZH2), which is a known component of polycomb repressive complex 2 (PRC2) that is able to triple-methylate H3K27. 117 Moreover, the transcription factor Hes family BHLH transcription factor 1 (HES1) is upregulated in UM cell lines due to H3K4 trimethylation of its promoter, predicting a higher risk for metastasis. 118 Di- and trimethylated H3K9 silence gene expression. The overall levels of H3K9me2 in melanoma tissues are higher than those in normal skin, which is mainly regulated by euchromatic histone lysine N-methyltransferase 2 (EHMT2/G9A).119,120 In several cancer subtypes, inhibition of EHMT2 led to the arrest of tumor growth. 121
Immune privilege and immune surveillance
Approximately 90% of UMs involve the choroid, and the remaining 10% occur in the iris or the ciliary body.122,123 The eye, especially the anterior chamber, including the iris and ciliary body, is considered ‘immune privileged,’ where immune responses to antigens are repressed to protect normal ocular tissues that would otherwise be damaged by excessive inflammation. The ocular ‘immune privileged’ compartment is achieved by anatomical and biochemical barriers.124,125 Anatomical barriers include the lack of afferent lymphatics and the existence of blood–ocular barriers that limit the access of systemic priming immune cells to the eye. Biochemical barriers include soluble immune suppressors, including cytotoxic T-lymphocyte antigen (CTLA)-2α, transforming growth factor beta (TGF-β), α-melanocyte-stimulating hormone, retinoic acid, and indoleamine dioxygenase (IDO), which inhibit the effect of immune cells that migrate to the eye. These immune suppressors can limit T-cell proliferation and convert T effectors into T-regulatory (Treg) cells. 126
Hence, UM may take advantage of this ‘immune privileged’ environment and allows immune-suppressive mechanisms to grow. There is evidence showing that large UM lesions heavily infiltrated by macrophages and CD8+ T cells harbor a genetic profile, which represents increased risk for liver metastases. 127 Even if CD8+ T cells infiltrate the tumor, UM can grow progressively in the eye globe,128,129 suggesting regional immune suppression. Several immune-suppressive mechanisms have been discovered, such as those involving programmed cell-death ligand-1 (PD-L1) and indoleamine 2,3-dioxygenase 1 (IDO1), to be utilized by UM to escape immune surveillance and metastasize.130–132 Primary UM cells showed resistance to CD8+ T cell cytolytic activity by expressing soluble Fas ligand (FasL), which provides protection from FasL-induced apoptosis.133,134 Moreover, UM is resistant to natural killer (NK) cell responses via expressing migration inhibitory factor (MIF) and TGF-β2.135–137 Recent studies have also shown that the metastasis of UM may be correlated with CD4+ Treg cells within lesions, which further supports a role for CD4+ Treg in tumor progression.138,139 In the mouse model of spontaneously developing UM, tumor dormancy is regulated in part by CD8+ T cells. 140 NK cells have been demonstrated to regulate the outgrowth of liver micrometastases in other intraocular melanoma mouse models.141,142
Thus, the evidence indicates that limited immune surveillance in the ‘immune privileged’ eye enables the UM tumor to be dormant, as patients often experience metastasis or recurrence after more than 5 years of a recurrence-free period.
Therapeutic options
Ocular treatment aims to conserve the eyeball and preserve visual performance. The treatment of primary tumors includes radiotherapy, phototherapy, and local resection, as well as reservation of enucleation in advanced cases. At present, no effective treatment exists to prevent metastasis, but early detection and intervention could be critical for a positive long-term survival outcome in UM.143,144 Metastatic UM responds poorly to chemotherapy, targeted therapy, and immunotherapy. Nevertheless, unlike cutaneous melanoma, UM metastases generally do not respond to immune-checkpoint inhibitors. However, novel therapeutics targeting mutant GNAQ/GNA11, regulators of G-protein signaling, downstream pathways, inactivated BAP1, and immune-checkpoint blockade are emerging.
Targeting oncogenic G-protein signaling: GNAQ/GNA11, PLCβ, CYSLTR2
As mutations in GNAQ and GNA11 are present in 80~93% of UM patients, to identify an effective agent targeting Gαq or Gα11 is a compelling approach to cure this malignancy. To date, the only published inhibitors of Gαq/11 subunits are a series of natural products represented by YM-254890 and FR900359. 145 YM-254890 was first derived from Chromobacterium spp. QS3666a, and used as a cyclic peptide to inhibit ADP-dependent platelet aggregation. 146 Mechanically, Gα subunits have two independent domains, the α-helical domain and the Ras-like domain, which are crucial for GDP–GTP exchange. The detachment of GDP and the following binding of GTP require the interface between the two domains. YM-254890 could bind to the hinge between the two domains, thus preventing domain opening and thereby preventing the exchange of GDP for GTP, leading to G-protein inhibition.147–149 YM-254890 preferably inhibits Gαq/11-mediated signaling, but not Gαi-, Gα15-, Gαs-mediated signaling, and the initiation of intracellular Ca2+ mobilization by PLCβ and Ca2+ channels. 146 However, YM-254890 might be effective, but only for R183 mutants of Gαq/11 in UM, which allows 40% of the GTP to continue to be bound, reflecting presumably a remaining activity of GTP hydrolysis. YM-254890 inhibited the accumulation of IP1 in cells expressing Gαq/11R183C, but had only a modest effect on Gαq/11Q209L, which is GTPase-deficient.39,45,146 This selectivity profile could be related to the distinct locations of two mutations relative to YM-254890 and/or to differences in the mechanisms by which G-protein function is modulated. 146 Recently, FR900359, a cyclic depsipeptide isolated from the Ardisia crenata Sims plant, was identified as a structural analog to YM-254890 that functions in a similar way. 39 It has been shown in vitro to specifically inhibit Gαq/11-mediated signal transduction in a human melanoma cell line that carries the Gαq/11Q209L mutation, resulting in decreased levels of PLC activity, G-protein subunit dissociation, and cellular responses to GPCR activation. 39 In UM92.1 and Mel202 cell lines, FR900359 could induce cell-cycle arrest and apoptosis via inhibiting oncogenic Gαq/11 signaling. In addition, FR900359 has been shown to promote Gαq/11Q209L-driven UM cell differentiation by reactivating PRC2-mediated gene silencing.150,151 Onken et al. have demonstrated that FR900359 suppresses nucleotide exchange and drives constitutively active Gαq/11Q209L into a quiescent GDP-bound state. 150 So far, only FR900359— but not YM-254890— has shown reasonable efficacy in Gαq/11Q209L-driven signaling. The kinetic parameters of the direct interaction between FR900359 and Gαq/11 have been determined recently. FR900359 dissociates from Gαq/11 with a remarkably slower off rate than that of YM-254890. 152 These findings suggest that FR900359 might be a useful agent for UM treatment. Although YM-254890 and FR900359 showed promising potential in inhibiting abnormally activated signaling in Gαq/11-mutant UM cell lines, they could not distinguish between wildtype and mutated Gαq/11, which added uncertainty to their potential clinical application. For instance, FR900359 suppressed ERK1/2 and AKTS473 phosphorylation selectively in cells carrying Gαq/11 mutations but reduced tonic IP1 levels in both Gαq/11-mutant and wildtype cells. 153 Besides, the complex structure and difficulties in the synthesis or production of these natural products have impeded their commercial development. Moreover, the exact pharmacological efficacy in UM patients, as well as the possible side effects still need further investigation. Development of FR900359 certainly provides the rationale for discovering effective agents targeting Gαq and Gα11, which is based on the understanding of the structure and transformation of these protein complexes. Identification and targeting of Gαq/11 interacting factors might be a novel approach to inhibit Gαq/11 activation in the future.
Apart from targeting the GDP–GTP exchange activity of Gα, there are also a few promising strategies used to target pharmaceutically Gα downstream effectors. As described above, the best understood Gα/effector interaction is that with PLC-β isozymes and diffuse B-cell lymphoma (Dbl) family proteins p63RhoGEF and Trio.59,154,155 Both bind to Gαq in a very similar pattern: a continuous helix-turn-helix (HTH) substructure of the effectors engages Gαq within its classical binding site, which consists of a groove formed between switch II (the G3 motif of the Ras-like domain) and helix α3. The direct interaction between the above effectors and Gα11 has not been confirmed. Understanding the binding mode of Gα subunits with their effectors has facilitated the development of compounds to antagonize the interaction of these signaling complexes. 156 The rearrangement of the switch regions of Gα subunits creates a hydrophobic cleft between switch II and helix α3 that is a major site of interaction with effectors and is the site for YM-254890 and FR900359 drug targeting. 145 Otherwise, the downstream effectors could engage the hydrophobic cleft of Gαq using their HTH substructures. The HTH of p63RhoGEF and PLC-β forms the interface with Gαq in crystal structures; the secondary interactions rearrange the complexes at membranes to activate effectors.157,158 Based on this structure, a linear peptide was designed and found to bind to PLC-β3 or p63RhoGEF in vitro by specifically interacting with activated Gαq, preventing recruitment and activation of downstream effectors. 156 It has no affinity for either GDP-bound Gαq or other G subunits, so it might be useful for inhibiting signaling cascades controlled by Gαq. Meanwhile, microinjection of the HTH-based peptide into mouse prefrontal cortex neurons can prevent downstream depolarization induced by muscarinic cholinergic receptor-dependent Gαq. 156 Inspired from the therapeutic options of targeting PLC-β3 and p63RhoGEF, strategies like a broad-spectrum peptide library should be applied to discover analogous peptides that serve as effector antagonists. Newly found Gαq/11-binding peptides might serve as leads for therapeutic development or provide effective affinity probes in the thermal shift assay of compound libraries to identify useful small molecules.
Another approach to targeting oncogenic G-protein signaling is the development of specific inhibitors of mutant GPCRs, such as those carrying CysLT2RL129Q, which is found in 3% of UM cases. A receptor-specific inverse agonist might be used to bind to and stabilize the inactive conformation of the receptor that could no longer activate Gαq/11. As a member of the same family, CysLT1R is shown to be highly expressed in colorectal adenocarcinomas, astrocytoma, ganglioglioma, and metastatic adenocarcinoma.159,160 CysLT1 antagonists, montelukast, zafirlukast, and pranlukast, are useful in the treatment of asthma and allergic rhinitis. Montelukast and pranlukast reduce colorectal tumor growth both in vitro and in vivo, via a combination of anti-proliferative and pro-apoptotic effects.161,162 BAY u9773 is a non-selective antagonist at both CysLT1 and CysLT2 receptors, whereas BayCysLT2 and HAMI 3379 are described as potent and selective CysLT2 antagonists.163–165 These two CysLT2R antagonists act as neutral antagonists, as both result in the reduction of CysLT2R-L129Q signaling, but they have limited efficacy as inverse agonists that target the oncogenic CysLT2R-L129Q mutant. Up to now, no studies have investigated the anti-cancer efficacy of specific CysLT2 antagonists in UM animal models or patients.
Targeting RGS
The inactivation process of Gαq/11 is catalyzed by the GAP function of RGS proteins. 166 Berstein et al. have first reported that the cycle of nucleotide exchange could be modulated by Gα-binding partners by demonstrating that PLC-β1 increases the activity of GTP hydrolysis by Gαq/11. Thus, PLC-β1 is both an effector and a GAP for Gαq/11 to exert paradoxical roles. The first evidence of GAPs came from a yeast-based genetic screen for mutants that elevated sensitivity of Saccharomyces cerevisiae to α-factor pheromone, which resulted in the identification of the two primary factors supersensitive-1 (Sst1) and supersensitive-2 (Sst2). 167 Sst1 and Sst2 rendered yeast supersensitive to α-factor. The large family of RGS proteins has a nine-α-helix bundle, which binds most covetously to the Gα transition state for GTP hydrolysis (GTP → GDP + Pi). 168
As RGS proteins are negative regulators of GPCR-mediated signaling, they are attractive targets for developing therapeutics. The development of RGS specific small molecules is still in its infancy, yet the ‘druggability’ of RGS domains has been determined early based on observations from the first crystal structure of the RGS4/Gαi1 complex. 169 It is hypothesized that a small molecule binding to the A-site on the RGS domain could theoretically block the interaction with Gα. Thus, it might be equally feasible to design a small molecule to allosterically improve the GAP function of endogenous RGS proteins.
Targeting second messengers: ARF6
ARF6 is a novel downstream effector of Gαq signaling, which transmits GNAQ as well as β-catenin signaling from the plasma membrane to the nucleus and cytoplasmic vesicles. It acts as an immediate downstream effector of GNAQ/GEP100 complex and delivers all the oncogenic signaling pathways of activated GNAQ, including PLC/PKC, Rho/Rac, YAP, and β-catenin. ARF-GEF inhibitors have been used as surrogates for ARF6 inhibition. To identify chemically tractable allosteric ARF6 inhibitors, Yoo et al. have performed a high-throughput screen with a collection of approximately 50,000 compounds. 71 They identified NAV-2729, a pyrazolopyrimidinone compound, as the most promising candidate. It has low micromolar potency with half maximal inhibitory concentration (IC50) value of 1.0 µmol/l determined using fluorometric ARF6 nucleotide exchange assays. Nevertheless, NAV-2729 showed high selectivity toward all other human ARF family members, at concentrations up to 50 µmol/l. NAV-2729 binds to ARF6 in the GEF-binding domain, instead of the nucleotide-binding pocket. NAV-2729 exhibited a spectrum of biological activities in UM cells. Treatment of UM cells with NAV-2729 reduced colony growth and also blocked all the downstream signaling pathways of GNAQ. 71 Altogether, these findings highlight ARF6 as a valuable therapeutic target for UM.
Targeting pathways: MAPK/MEK/ERK, PKC, PI3K/Akt/mTOR, Trio/Rho/Rac/YAP
MAPK is constitutively activated in nearly 90% of metastatic UM. In experimental studies, several MEK1/2 inhibitors such as trametinib, selumetinib, and PD0325901, induced growth arrest in GNAQ/GNA11-mutant UM cell lines, UM cell-line-derived xenograft, and patient-derived xenograft (PDX) models.170–173 However, clinical studies have shown variable outcomes for MEK inhibition. A preclinical study has demonstrated that the five UM cell lines used had GNAQ or GNA11 mutations and were either moderately or highly sensitive to the MEK inhibitor TAK733, with IC50 values below 10 nmol/. 174 A phase I study of TAK-733 on 12 UM patients in total 51 patients with advanced solid tumors showed a limited antitumor effect; only two patients with cutaneous melanoma (one with BRAF mutation) had partial responses [ClinicalTrials.gov identifier: NCT00948467]. 175 In a phase II trial with 120 advanced UM patients, selumetinib improved the median PFS to 15.9 weeks compared with only 7 weeks for chemotherapy, but only modestly increased the median OS [ClinicalTrials.gov identifier: NCT01143402]. 176 Another phase I trial of selumetinib is still recruiting metastatic UM patients to examine whether higher drug dose efficiently blocks the MAPK pathway and prevents resistance [ClinicalTrials.gov identifier: NCT02768766]. Several studies have investigated the beneficial effects of the combinations of MEK inhibitors with other drugs. In the phase III clinical trial, SUMIT, involving 129 metastatic UM patients, the combination of selumetinib and dacarbazine failed to improve PFS, with a reported low response rate of 3.1% and no statistically significant benefit in OS [ClinicalTrials.gov identifier: NCT01974752]. 177 A PDX model-based investigation has been done in five UM PDXs, and showed that the combinations of selumetinib with mTORC1/2 inhibitor vistusertib (AZD2014) and ERK inhibitor AZ6197 exerted the best activity, as tumor growth inhibition (TGI) was 62–97% for the five PDXs in the selumetinib + AZ6197 group, and 59–83% for the five PDXs in the selumetinib + AZD2014 group. The TGI value of monotherapies was 11–34% for selumetinib, 0–67% for AZ6197, and 28–84% for AZD2014. An objective response rate (ORR) below −0.5 was achieved in all five models for both combination groups. 178 Recently, a review of 590 cases from six eligible clinical studies has shown that UM is poorly responsive to MEK inhibitors, including selumetinib [median PFS 16 weeks, median OS 11.8 months, 14% partial response (PR), 1-year OS rate 45% 176 ], trametinib (median PFS 1.8 months, ORR/PR/complete response 0% 179 ), and combined applications (selumetinib + dacarbazine: median PFS 2.8 months, 1-year OS rate 50%, ORR 3%; 180 trametinib + AKT inhibitor uprosertib: median PFS 15.7 weeks; 181 binimetinib + PKC inhibitor sotrastaurin: median PFS 3.1–4 weeks) [ClinicalTrials.gov identifier: NCT01801358]. 182 A recent three-arm randomized phase II study has demonstrated a statistically significant improvement in PFS for metastatic UM, from 3.4 months for selumetinib alone to 4.8 months for selumetinib in combination with paclitaxel (PT), without a significant increase in toxicity [ISRCTN 29621851]. 183 For cutaneous melanoma, which has been treated with BRAF inhibitors, the combination of MEK and BRAF inhibitors benefited patients who had metastatic melanoma with BRAF-V600E or -V600K mutations. In a phase III trial, the combination of trametinib and dabrafenib, compared with dabrafenib alone, improved the PFS of patients from 8.8 months to 9.3 months. 184 The MEK inhibitor and BRAF inhibitor combination has not been tested in UM patients, as activating BRAF mutations are usually absent in UM. GNAQ/11 is the upstream regulator of the RAF/MEK/ERK pathway, and also activates other cascades such as the PI3K/Akt/mTOR and Trio/Rho/Rac/YAP, which makes the blocking of the RAF/MEK/ERK pathway inappropriate for UM. Regarding UM, due to the lack of GNAQ/11 inhibitors, such drug combinations need further research and development.
The poor response of UM patients to MEK inhibitors may be partially due to the complex tumor microenvironment. Targeting of c-mesenchymal-epithelial transition factor (c-MET), the receptor of hepatocyte growth factor (HGF), enhanced the effects of trametinib in metastatic UM. HGF is secreted in the liver and phosphorylated c-MET is detected in UM liver metastases. This suggests that HGF induces resistance of tumor cells to trametinib.172,185 Activation of the PI3K/Akt pathway by HGF might be another resistance mechanism, which might be reversed using the PI3K inhibitor GDC0032. 185 These findings support a previous study, which has reported the effective combination of PI3K and MEK inhibitors for UM.172,173
Inhibition of PKC alone with AEB071 (sotrastaurin) unsustainably suppressed ERK1/2 signaling and induced cell-cycle G1 phase arrest. 171 In a phase I clinical trial, 4 (3%) of 153 metastatic UM patients had a partial response and 76 (50%) had stable disease. Tumor reduction by ⩾10% from baseline was observed in 34 patients (22%) [ClinicalTrials.gov identifier: NCT01430416]. 186 This effect was enhanced by the combination with MEK inhibitors, MEK162 and PD0325901, as shown in an in vivo animal study. 171 However, a phase Ib/II clinical trial that combined the MEK inhibitor binimetinib with AEB071 was terminated prior to initiation of the phase II trial [ClinicalTrials.gov identifier: NCT01801358]. In addition, the combination of AEB071 with the p53-MDM2 inhibitor CGM097 or the mTORC1 inhibitor RAD001, showed promising results of tumor regression. AEB071 + RAD001 co-treatment induced significant tumor regression in two of the three PDX models, while the AEB071 + CGM097 combination led to tumor regression or stasis in all five PDX models. 187 Another novel PKC inhibitor, LXS196, is being assessed for its safety, tolerability, pharmacokinetics (PK), pharmacodynamics (PD) and efficacy in 68 patients with metastatic UM. Among patients treated with LXS196, 6 had PR and 45 had stable disease (SD) as their best response, suggesting promising clinical activity for LXS196 as a single agent with manageable toxicity profile [ClinicalTrials.gov identifier: NCT02601378]. 188
The combination of the pan-PI3K inhibitor GSK2126458 and the MEK inhibitor GSK1120212 induced apoptosis in GNAQ/11-mutant UM cell lines. 173 The combination of AEB071 with the PI3K inhibitor BYL719 showed a synergistic effect in mouse models with UM cell-line-derived subcutaneous transplantation. 189 Following up from this study, a phase I clinical trial is currently underway to determine whether simultaneous inhibition of PI3K and a different downstream pathway could improve the anticancer effect in metastatic UM [ClinicalTrials.gov identifier: NCT02273219].
The combination of selumetinib with the Akt inhibitor MK2206 induced synergistic effect on autophagic death of UM cells in vitro and in vivo (cell-line-derived subcutaneous transplantation). 170 However, a phase II clinical trial of trametinib combined with the Akt inhibitor GSK795 showed no improvement of PFS and response rate (RR) compared with that in the selumetinib group [ClinicalTrials.gov identifier: NCT01979523]. 181
Regarding mTOR-targeting therapy, a combination screening showed a potent synergetic interaction between the PI3K inhibitor GDC0941 and the mTOR inhibitor everolimus in vitro and in two PDX models. 190 A clinical phase II study using the combination of everolimus and the somatostatin receptor agonist pasireotide showed poor benefit and required dose reduction [ClinicalTrials.gov identifier: NCT01252251]. Overall, 3 of 13 (26%) patients obtained clinical benefit, 7 of 13 (54%) demonstrated SD, and 7 of 14 (50%) required at least one dose reduction due to toxicity. 191
The Trio/Rho/Rac/YAP pathway has recently been found to play a vital role in GNAQ/11 downstream signaling. 192 The YAP inhibitor verteporfin decreased the growth of GNAQ/11-mutated UM cells.62,63,193 In addition, Gαq activates focal adhesion kinase (FAK), which facilitates YAP activation. Inhibition of FAK activity by VS-4718 or PF562771 blocks YAP signaling and tumor growth, indicating FAK is an actionable target in UM. 192
The molecular targets and potential drugs are summarized in Tables 1 and 2. It is not surprising that these compounds often show limited effects in clinical trials, as they only affect part of the oncogenic Gα11/q networks. A thorough understanding of the signaling pathways orchestrated by Gα11/q combined with new target-specific drugs will certainly promote the progress of UM therapy.
Molecular targets and potential drugs of UM in preclinical studies.
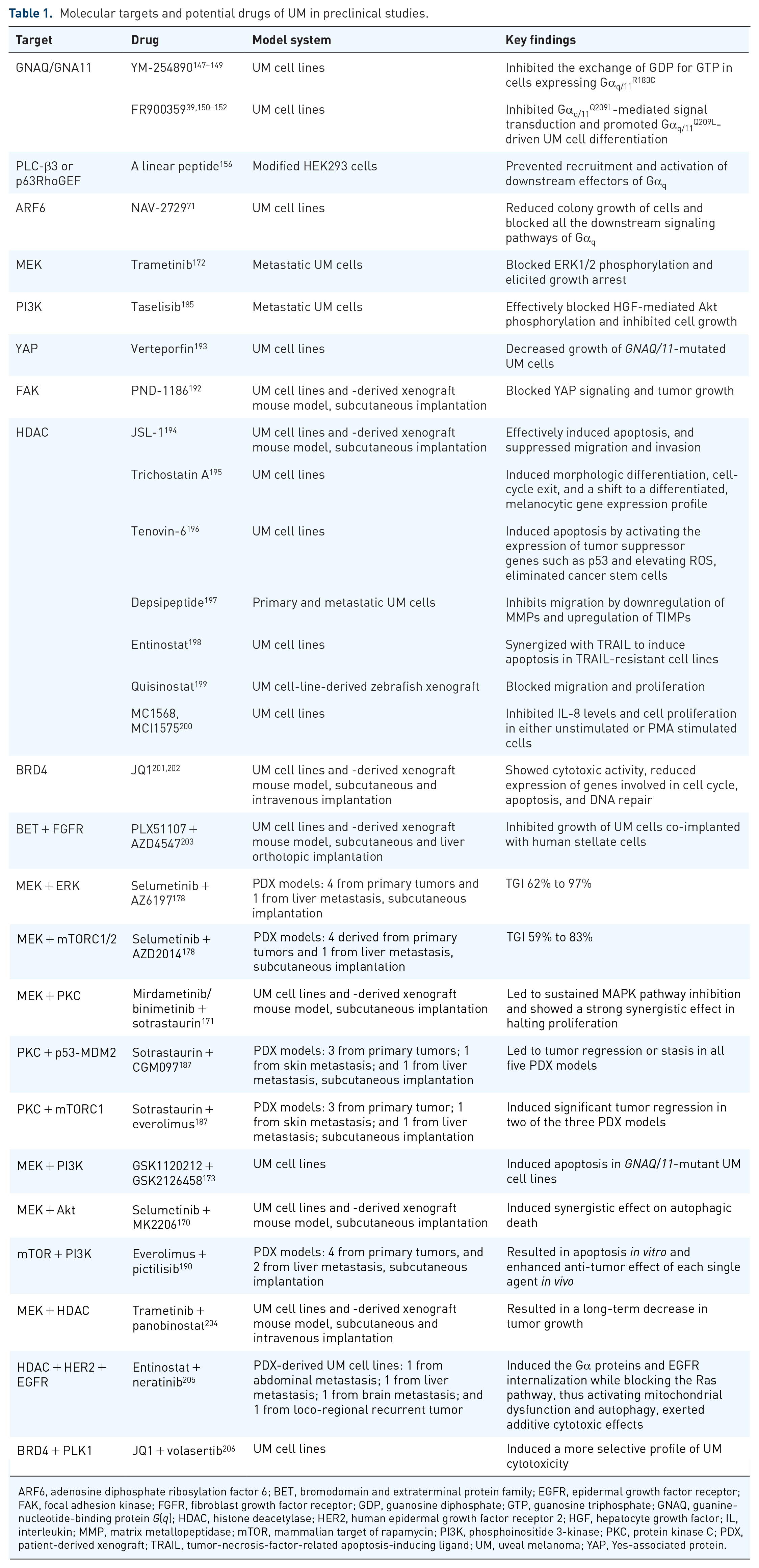
ARF6, adenosine diphosphate ribosylation factor 6; BET, bromodomain and extraterminal protein family; EGFR, epidermal growth factor receptor; FAK, focal adhesion kinase; FGFR, fibroblast growth factor receptor; GDP, guanosine diphosphate; GTP, guanosine triphosphate; GNAQ, guanine-nucleotide-binding protein G(q); HDAC, histone deacetylase; HER2, human epidermal growth factor receptor 2; HGF, hepatocyte growth factor; IL, interleukin; MMP, matrix metallopeptidase; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinase; PKC, protein kinase C; PDX, patient-derived xenograft; TRAIL, tumor-necrosis-factor-related apoptosis-inducing ligand; UM, uveal melanoma; YAP, Yes-associated protein.
Molecular targets, potential drugs and ongoing clinical trials for UM.

The information of these unfinished clinical trials is available at www.clinicaltrials.gov.
CTLA-4, cytotoxic T-lymphocyte-associated antigen 4; GDP, guanosine diphosphate; GTP, guanosine triphosphate; HDAC, histone deacetylase; ImmTAC, immune-mobilizing monoclonal T-cell receptor against cancer; mTOR, mammalian target of rapamycin; ORR, objective response rate; OS, overall survival; PFS, progression-free survival; PI3K, phosphoinositide 3-kinase; PR, partial response; PLC-β3, phospholipase-C beta 3; PD-1, programmed cell-death 1; PKC, protein kinase C; RR, response rate; SD, stable disease; UM, uveal melanoma.
BAP1: HDAC inhibitor, BET inhibitor
BAP1 loss in class 1 UM led to H2A hyperubiquitination, which could be reversed by inhibition of histone deacetylases (HDACs), which reprogram aggressive UM to a highly differentiated and low-grade phenotype in vivo. 195 HDACs are a class of epigenetic enzymes that remove acetyl groups from acetylated lysine residues of histone proteins. Histone acetylation is related to the regression of gene transcription, including different classes of cancer suppressor genes. Several HDAC inhibitors have manifested promising anticancer activities, such as valproic acid, panobinostat, vorinostat, 195 trichostatin A,195,212 tenovin-6, 196 depsipeptide, 197 MS-275, 198 quisinostat, 199 JSL-1, 194 MC1568, and MCI1575. 200 For example, JSL-1, a novel HDAC inhibitor, effectively induced apoptosis, and suppressed the migration and invasion of UM cells and UM growth in a cell-line-derived xenograft mouse model. 194 JSL-1 impaired the self-renewal capacity and eliminated stem-like cells which are believed to be seeds of metastasis. A recent preclinical research identified HDAC inhibitors as potential candidates that suppress the adaptive YAP and Akt signaling following MEK inhibition. The MEK–HDAC inhibitor combination outperformed either agent alone, resulting in a long-term decrease in tumor growth in both subcutaneous and liver metastasis models. 204 In addition, Booth et al. have used PDX-derived UM cell lines to confirm that combining the HDAC inhibitor entinostat with neratinib, an inhibitor of human epidermal growth factor receptor 2 and epidermal growth factor receptor (EGFR) tyrosine kinases, exerts additive cytotoxic effects. This combination cooperatively induced the Gα proteins and EGFR internalization while blocking the Ras pathway, thus activating mitochondrial dysfunction and autophagy. 205
An ongoing phase II trial is examining the efficacy of the HDAC inhibitor vorinostat in patients with metastatic UM [ClinicalTrials.gov identifier: NCT01587352]. In addition, a multicenter phase II open label study evaluating the effect of the combination of the anti-PD-1 pembrolizumab and entinostat showed clinical efficacy in metastatic UM patients with manageable toxicities, which might be attributed to the potential immune-modulatory activity of HDAC inhibitors. The ORR was 10% and 31% of patients exhibited the best overall response of SD [ClinicalTrials.gov identifier: NCT02697630].209,210 With the development of fine needle aspirate biopsies, another study assessed whether using vorinostat in class 2 high-risk UM patients could switch their gene expression profile into normal melanocytes [ClinicalTrials.gov identifier: NCT03022565]. 195
Apart from H2A hyperubiquitination, global DNA methylation is another important consequence of BAP1 loss, revealed by multiplatform analysis of 80 primary UM. 22 The DNA methylation gene profile is different between EIF1AX- and SRSF2/SF3B1-mutant UM. Class II or BAP1-mutant UM showed a unique phylogenetic cluster of global DNA methylation. 213 These findings indicated that the DNA methylation status, as well as other mechanisms of regulation of gene transcription, might be vital in regulating metastasis in UM. The bromodomain containing 4 (BRD4) inhibitor JQ1 showed cytotoxic activity in UM cells and cell-line-derived xenograft mouse models bearing GNAQ/11 mutations. It reduced the expression of genes involved in cell cycle, apoptosis, and DNA repair.201,202 BRD2, BRD3, BRD4, as well as bromodomain testis-associated (BRDT) proteins belong to the bromodomain and extraterminal (BET) protein family, which promotes transcriptional elongation via binding to acetylated lysine of histones and recruiting transcriptional complexes.214,215 JQ1 treatment of UM cells made them more sensitive to cell-cycle-related inhibitors such as the Polo-like kinase 1 (PLK1) inhibitor BI6727 volasertib. 206
PLX51107, a second-generation BET inhibitor, is undergoing clinical trial in patients with advanced UM, as well as other cancers [ClinicalTrials.gov identifier: NCT02683395]. 216 However, cases with metastases manifested resistance to PLX51107. The combination of the fibroblast growth factor receptor (FGFR) inhibitor AZD4547 with PLX51107 inhibited the growth of UM cells co-implanted with human stellate cells, indicating that the concomitant inhibition of the BRD4 and FGFR pathways might be a novel option for UM liver metastases. 203
The use of HDAC and BET inhibitors might lead to a less aggressive and actively differentiated state of BAP1-deficient UM cells, therefore prolonging the survival of UM patients. As expected, with the emergence of more and more epigenetic drugs, the combination of HDAC or BET inhibitors with other therapies may become an active research topic in UM therapy.
Immune-checkpoint blockade: CTLA-4, PD1/PD-L1
The poor efficacy of immune-checkpoint blockade suggests that melanomas arising from the uveal tract might be immunotherapy-resistant variants. Ipilimumab, a monoclonal antibody targeting cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), has shown an RR ranging from 0% to 5% and a median OS of 5.2–10.3 months [ClinicalTrials.gov identifier: NCT00495066]. 207 A single-arm phase II study of pembrolizumab in patients with metastatic UM obtained a median PFS at 11 months, with 20% RR and 60% clinical benefit rate. 208 The largest retrospective study involving 58 metastatic UM patients using anti-PD-1/PD-L1 immunotherapy has shown an objective RR of 3.6%, median PFS of 2.6 months, and median OS of 7.6 months. 217 A study in the Netherlands has shown that 2 of 15 patients who received at least 1 anti-PD1 therapy had clinical benefit. Two patients were alive, and on treatment showed SD. The median OS was 9.6 months, and PFS was 2.3 months. 218 The weak efficiency of immunotherapy might be attributed to a very low mutational load of UM and liver being an immunosuppressive organ.219–221 Analysis of eight cases enrolled in a single-center trial indicated that metastatic UM patients treated with the combination of nivolumab and ipilimumab through transarterial chemoembolization (TACE) had a 25% PR rate and 50% SD rate. 222 A largescale phase II trial, GEM1402, was conducted with the combination of ipilimumab + nivolumab in a group of 50 metastatic UM patients. The ORR and SD rates were 12% and 52%, respectively [ClinicalTrials.gov identifier: NCT02626962]. 211 A more recent phase II clinical trial enrolled 39 metastatic UM patients who received nivolumab plus ipilimumab followed by nivolumab maintenance. The best ORR was 17% for PR, 53% for SD, and 30% for progression of disease. The median PFS and OS was 26 weeks and 83 weeks, respectively, and the 1-year OS was 62% [ClinicalTrials.gov identifier: NCT01585194]. 223 Compared with cutaneous melanoma patients, the immunotherapy results of UM patients were less optimistic. As cutaneous melanoma shows a higher mutational burden than UM, which is related to a large number of neo-antigens, it is suitable for immunotherapy.
Previously, Nitta et al. have identified an active T-cell repertoire comprising tumor-infiltrating lymphocytes (TILs) in UM patients. 224 In a phase II two-stage study, 21 metastatic UM patients were enrolled. Administration of a single infusion of TILs induced objective tumor regression in 7 of 20 evaluable (35%) patients. Among them, one patient with highly pretreated UM manifested a durable complete regression of all the metastatic lesions, which has been going on for almost 2 years now. 225
Tebentafusp is a novel technique of immunotherapy based on the immune-mobilizing monoclonal T-cell receptor against cancer (ImmTAC). It contains a soluble T-cell receptor HLA-A*02:01, in complex with a melanocyte-lineage-specific antigen gp100280–288, and is fused to an anti-CD3 single-chain variable fragment. 226 The expression levels of Ggp100 are higher in melanoma cells compared with normal melanocytes and other tissues. 227 Preclinical data show that tebentafusp induces the formation of an immune synapse between T cells and tumor cells to cause cytolysis, while it induces the production of a range of pro-inflammatory cytokines including TNFα, interleukin 2 (IL-2), IL-6 and interferon-γ.226,228,229 According to the results from two completed clinical trials with tebentafusp in metastatic UM patients, the ORR, median PFS, and OS rates were 14–18%, 3.7–5.6 months, and 73–74% respectively. The adverse events of tebentafusp were transient and manageable [ClinicalTrials.gov identifiers: NCT01211262; NCT02570308; phase I]. These reports on tebentafusp in metastatic UM are encouraging, although the number of patients enrolled was small (n = 34).230,231 There are two more ongoing trials for tebentafusp in UM [ClinicalTrials.gov identifiers: NCT03070392, NCT02570308; phase II].
Liver-directed therapies
Liver-directed targeted therapies are being pursued for the hepatotropic feature of metastatic UM. 94 The presence of multiple liver metastases is a contraindication for surgical excision, and thus only a small number of cases are eligible for surgical treatment. 232 In cancer management, embolization blocks the blood supply to the tumor, and often includes an ingredient to attack the tumor chemically or with irradiation. TACE is the usual form. 233 Chemoembolization is a method of local chemotherapy that combines infusion of chemotherapeutic drugs through the hepatic artery with the blockage of blood supply to the tumor. The chemotherapeutic drugs include carboplatin alone, cisplatin alone or in combination with carboplatin, and mitomycin C alone or in combination with doxorubicin and cisplatin.234,235 Immunoembolization means infusion of an immune-stimulating agent, such as granulocyte–macrophage colony-stimulating factor (GM-CSF), into the hepatic artery, followed by embolization. It acts as a stimulus to the immune system against tumor cells. 236 Radioembolization using yttrium-90 ( 90 Y) has been used to treat liver metastases from UM. In a phase II trial, radioembolization was performed with 90 Y resin microspheres, and its effectiveness was calculated using a specific formula. The median OS and PFS was 10 months and 4.7 months, respectively. 237 A phase I study of radioembolization in combination with ipilimumab and nivolumab for metastatic UM is underway [ClinicalTrials.gov identifier: NCT02913417]. Isolated hepatic perfusion (IHP) is a procedure by which the liver is surgically isolated and perfused with a chemotherapeutic agent to allow local perfusion of the liver with a high dose of a chemotherapeutic agent. 238 A randomized phase III trial of IHP versus best alternative care (BAC) for metastatic UM is underway [ClinicalTrials.gov identifier: NCT01785316]. BAC implicates that the treating physician at each study center decides the treatment together with the patient, in consideration of all available regimens, such as surgery and other experimental treatments tolerated. 239 All of the above approaches are used to treat metastatic liver disease; however, there is no standard treatment available.
Conclusion
Although activation mutations in Gαq and Gα11 genes are dominant in UM, direct inhibition of the constitutively active oncoproteins Gαq and Gα11 is still in its infancy. Targeting separate downstream pathways by, for example, a MEK inhibitor has limited effects. Combinations of inhibitors of multiple signaling molecules and compounds targeting ARF6 might be sufficient to correct all the known downstream pathways. Modifiers of mutated BAP1 and epigenetics, such as HDAC and BET inhibitors, may become useful tools to revert the high-risk UM phenotype, since they have shown promising outcomes in preclinical models. Immune-checkpoint blockade has inadequate anticancer activity in systemic use, as the eye globe is ‘immune privileged’ and the liver maintains an immunosuppressive environment. TIL and tebentafusp, as well as liver-directed therapies, brought new hope for metastatic UM, which need to be further developed.
Despite great progress in the development of novel therapeutic strategies, UM, especially metastatic UM, remains an incurable malignancy. Compared with cutaneous melanoma, UM shows poor sensitivity to targeted therapies, such as immunotherapy and MEK inhibitors. This may be caused by the different biological mechanisms and behaviors of these two malignancies. Therefore, different therapeutic approaches are required. Further studies on the genetics, epigenetics, tumor microenvironment, and immunologic background of UM will help to discover effective personalized therapies.
An important lesson to bear in mind while searching for therapeutics is that UM’s biology and pharmacology are quite unique compared with other types of cancer. Research results from cell lines or xenograft models might only partially represent the whole picture of UM in vivo. This has been repeatedly manifested in the past by the fact that some agents that were effective in preclinical models completely failed in clinical trials. In addition, the results from PDX models should be interpreted with caution before testing the effectiveness of the new drugs in clinical trials, as the tumor microenvironment in UM patients is much more complicated than that of mice, especially that of the wildly used immune-deficient nude mice. These differences could result in unexpected outcomes, and humanized animal models should be considered.
In conclusion, UM therapy is largely an unmet medical need. Future investigations should address all altered genes, proteins, and dysregulated signaling networks in this disease at the system level to increase understanding and pave the way for personalized therapy. New discoveries in the field will allow for improvement in clinical outcomes.
Footnotes
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
The authors disclose receipt of the following financial support for the research, authorship, and/or publication of this article: this research was supported by the Science and Technology Commission of Shanghai (17DZ2260100, 19JC1410200), the National Natural Science Foundation of China (grants U1932135, 81772875, 81770961, 81802739), National Key Research and Development Plan (2018YFC1106100), Shanghai Municipal Education Commission—Gaofeng Clinical Medicine Grant Support (RC20190081) and Innovative research team of high-level local universities in Shanghai to J. Zhang. The funders played no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.