
Abstract
Cardiac tumors are rare and complex entities. Early assessment and differentiation between non-neoplastic and neoplastic masses, be they benign or malignant, is essential for guiding diagnosis, determining prognosis, and planning therapy. Cardiac sarcomas represent the most frequent primary malignant histotype. They could have manifold presentations so that the diagnosis is often belated. Moreover, considering their rarity and the limitation due to the cardiac location itself, the optimal multimodal management of patients affected by primary cardiac sarcomas still remains highly difficult and outcome dismal. Therefore, there is an urgent need to improve these results mainly focusing on more adequate tools for prompt diagnosis and exploring new and more effective therapies. Knowledge about the molecular landscape and pathogenesis of cardiac sarcoma is even more limited due to the rarity of this disease. In this sense, the molecular characterization of heart tumors could unfold potentially novel, druggable targets. In this review, we focused on genetic aberrations and molecular biology of cardiac sarcomas, collecting the scarce information available and resuming all the molecular findings discovered in each tumor subtype, with the aim to get further insights on mechanisms involved in tumor growth and to possibly highlight specific molecular profiles that can be used as diagnostic tests and unveil new clinically actionable targets in this tricky and challenging disease.
Keywords
Background
Cardiac tumors, both benign and malignant, are rare diseases. They account only for a small fraction of all cardiac masses, mostly represented by pseudo-tumors like thrombi, vegetations, and aneurysms. The majority of initial medical reports on cardiac tumors were based on autopsy findings. With echocardiography increasingly used since the 1960s, successful diagnosis of ante-mortem cardiac tumors has risen. Their estimated frequency ranges from 0.0017% to 0.33%, mostly represented by benign tumors (75%), with cardiac myxomas being the most common accounting for nearly half of them. 1 Among malignant cases, secondary tumors of the heart, either by metastatic spread or by direct invasion, are far more common than primary cardiac tumors. Primary tumors are mainly sarcomatous in nature, whereas metastases could originate from various organs, generally from lung. 2
Location, morphology, and extent of the mass provide important and meaningful information; half of the primary tumors of the right atrium are malignant while most left atrium tumors are benign. Other imaging findings suggestive of malignancy include involvement of more than one cardiac chamber, size >5 cm, hemorrhagic pericardial effusion, broad base of attachment, and extension into the mediastinum. 3 Early assessment and differentiation between non-neoplastic and neoplastic masses is essential for guiding diagnosis, determining prognosis, and planning therapy.
Whilst being extremely rare, sarcomas of the heart represent the most frequent primary malignant histotype (75–95%). Virtually all types of sarcomas may be observed in the heart with a predominance of angiosarcomas (AS) or undifferentiated sarcomas, while other sarcomas are less frequently described (leiomyosarcoma, rhabdomyosarcoma, and myxofibrosarcoma). 4 They could have clinical presentations ranging from cough and general fatigue to thromboembolism, pericardial effusion, arrhythmias, dyspnea, hemoptysis, or symptoms of congestive heart failure depending on tumor location and on pathological subtype. The tumor site can give an indication of the most probable tumor type in some cases, with the vast majority of AS and myxoma developing in the right atrium and left atrium, respectively.
Due to the rarity of the disease, evidence for the optimal multimodal management of primary cardiac sarcomas is limited. There is no standardized approach, which is then often extrapolated from that of soft tissues sarcomas (STS) in other sites, thus adopting a “one size fits all” approach. Moreover, inevitable limits due to the cardiac location itself must be considered. 5 Complete surgical resection provides the greatest chance for survival in primary localized cardiac sarcoma, but it is rarely achievable in practice given the aggressive and infiltrative nature of these tumors. Moreover, radiotherapy, an effective adjuvant tool for sarcomas of the extremities, is barely applicable to cardiac sarcomas considering that the heart itself is usually sensitive to radiation injury leading to substantial risk of cardiomyopathy and chronic pericarditis. Finally, the typical cardiotoxicity of anthracyclines, which represent the preferred chemotherapeutic choice for sarcomas, could limit their use in neoadjuvant/adjuvant or advanced settings in these cases.
Even if based on retrospective, small, and heterogeneous case series, sarcomas of the heart seem to be very aggressive tumors with a dismal prognosis; tumor recurrence and metastases represent frequent and often early events. A high rate of both relapse and distant metastases within the follow-up period is reported in a series of malignant cardiac tumors seen over 15 years (45.5% and 72.2%, respectively) 6 and the median overall survival (OS) is scanty: in older retrospective case series, most patients died within 12–16.5 months after initial diagnosis. 7 In more recent series, survival reaches 38.8 months but only for patients who underwent complete resection in referral centers. Long-term survivals are uncommon and significantly depend on the histotype. 4
Knowledge about the molecular genetics and pathogenesis of cardiac sarcoma is even more limited. Most publications consist of case reports, with only a few reporting clinical/pathological and molecular features of cohorts of cardiac sarcomas (Table 1).
Molecular analysis of primary cardiac sarcoma available in literature.

In this review, we focus on genetic aberrations and molecular biology of cardiac sarcoma, collecting the scarce information available and summarizing molecular findings of each tumor subtype with the aim of highlighting specific molecular profiles that may be used as diagnostic tests or potential targets for therapy in this complex and challenging disease.
Angiosarcoma
AS is the most common differentiated cardiac sarcoma, generally originating in the right atrium. It represents about 25–40% of cases of cardiac sarcoma, occurring more frequently in men within the third and fifth decade of life (Figure 1). 36
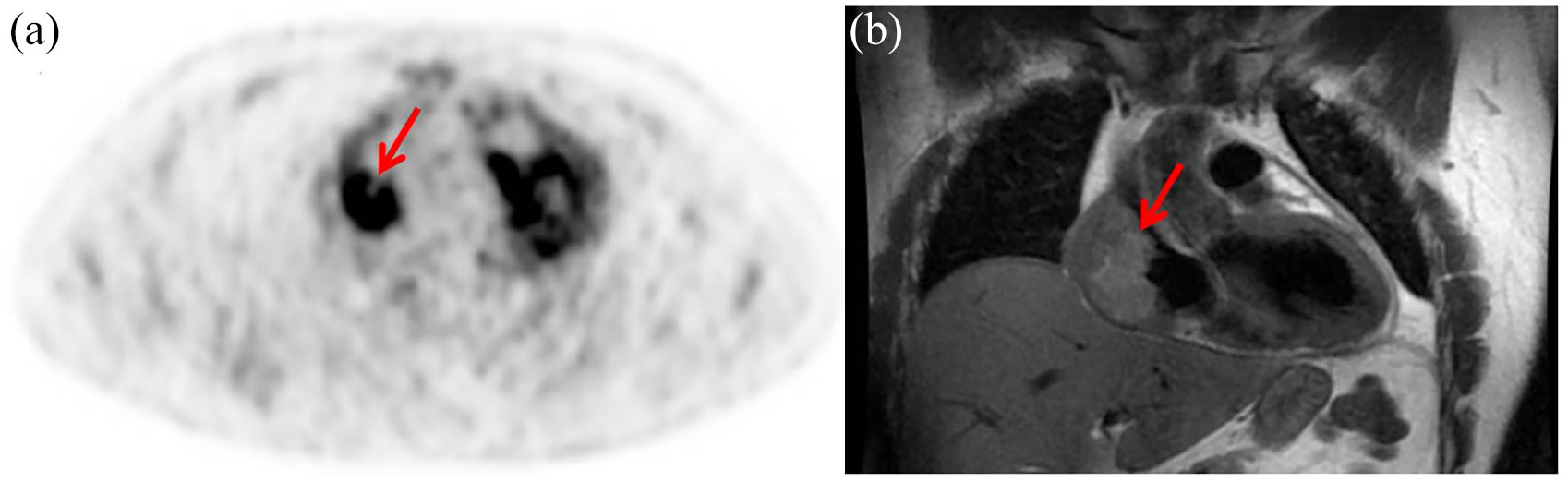
Angiosarcoma of the right atrium in a 38-year old man. 18F-FDG PET/CT shows abnormal uptake (SUVmax = 8.7) in the right atrium (a). FDG uptake corresponds to the voluminous (6 × 4.7 × 3.4 cm) neoplastic mass showed by the coronal image of the cardiac MRI (b). For clarity, the uptake visible on the left refers to the physiological FDG uptake due to the contractile activity of the left ventricle.
Cardiac AS is defined by endothelial differentiation and characterized by a high proliferation rate, permeating and destructive nature, propensity to metastasize, and poor prognosis with patients succumbing to disease within a few months of diagnosis. 37 Molecular aberrations detected in cardiac AS often involve KDR and KIT and homozygous deletion of CDKN2A.10,12,14
It has been shown that almost half of soft tissue AS carry recurrent somatic mutations in angiogenic signaling pathways, providing a rationale for investigating targeted therapies. In particular these mutations involve KDR (kinase insert domain receptor encoding for one of the vascular endothelial growth factor receptor tyrosine kinases), PLCG1 (phospholipase C gamma 1 encoding for a tyrosine kinase signal transducer within the phosphoinositide signaling pathway), and PTPRB (protein tyrosine phosphatase receptor type B encoding for a negative regulator of vascular growth factor tyrosine kinases).38,39
Indeed, KDR mutations are present in about 7–10% of soft tissue AS,38,40 and three KDR-mutated cases have been reported in cardiac AS: one case harbored a mutation in the transmembrane domain (p.T771K) while the other two in the immunoglobulin domain (p.G681R and p.R720W) of the protein.9,12 Similarly, PLCG1 mutations were reported in about 10% of soft tissue AS, 38 predominantly affecting the highly conserved auto-inhibitory Src homology 2 (cSH2) domain within exon 18 (p.R707Q or p.R707L), with rarer mutations involving exon 11.38,39 Among cardiac cases, p.R707Q has been reported in a minority of cases and functional studies demonstrate that this mutation confers KDR-independent PLCG1 activation, probably causing primary resistance against VEGF/KDR-directed therapies. 12 Interestingly, it seems that PLCG1 and KDR mutations are mutually exclusive in AS. 38 However, PTPRB mutations have yet to be reported in cardiac AS, although they are present in up to 26% of soft tissue AS, exclusively in the setting of secondary or MYC-amplified AS and often in association with PLCG1 mutations.38,39
Another frequent genetic alteration in soft tissue AS is MYC amplification. However, it is generally regarded as the hallmark of post-radiation malignancies including chronic lymphedema-associated AS, with only a small subset of primary AS harboring this abnormality. 38 Thus, this event is extremely rare in the cardiac setting, and only one case has been reported. 13
Alterations of TP53, commonly observed in sarcoma with complex karyotypes, are rare events in all AS, with an estimated frequency of 4%. 41 In cardiac AS, the exact frequency and significance of TP53 mutations remain unclear, given the limited literature on the subject. In the work of Naka et al., two mutated cases were reported 16 and another case was identified by Zu et al. 14 However, no TP53 mutation was detected in three cases analyzed by Garcia et al. 15 and neither in another seven cases analyzed through next generation sequencing approaches.9,12
Alterations of the RAS pathway seemed to have a role in AS onset: H/K/N-RAS mutated cases have been reported in about 13% of AS from various sites, 39 with KRAS mutations being relatively frequent (26%) in hepatic AS. 42 In cardiac AS, p.G13S and p.Q61K mutations have been reported in few cases.12,15
Other mutated genes detected in soft tissue AS (CIC fusions and mutations, PIK3CA, FLT4, TIE1) 38 have not yet been investigated in the cardiac AS. Conversely, some mutations have been identified only in cardiac AS and not in other soft tissue AS. For example, inactivating mutations of the lysine methyltransferase 2D (KMT2D), a gene found altered in a large number of different cancers (including diffuse large B-cell lymphoma, follicular lymphoma, and medulloblastoma), have been reported in cardiac AS but not in extra-cardiac AS: one in concomitance with KDR p.R707Q and one together with Protection of Telomeres 1 (POT1) inactivating mutation. 12
With regards to the latter, POT1 alterations occur in 27% of Li–Fraumeni-like (LFL) family with members affected with AS, and in 11.4% of sporadic cardiac AS. 8 Up to now, different POT1 mutations have been identified in cardiac AS: one p.G301* and 3 cases of p.R117C in LFL families, and a p.P116L and a p.R432* in two sporadic cases.8,11,12
Regarding chemotherapy, doxorubicin-based regimens remain the recommended first-line schemes for AS, as for other histological subtypes of STS. For second and further lines, no specific algorithm of treatment has been established. Among possible strategies for STS, taxanes have shown efficacy specifically in AS. In 1999, Fata et al. first reported an interesting rate of response to paclitaxel in patients with AS of the scalp or face in a small retrospective single-center study suggesting the potential role of taxanes for the treatment of advanced or metastatic AS. 43 Eight out of nine patients had major responses (four partial responses and four clinical complete responses) with a median duration of 5 months (range, 2–13 months). 43 Subsequently, this data was reinforced by a retrospective study on a larger number of patients and confirmed by the phase II trial ANGIOTAX.44,45 Results from 32 patients collected from 10 centers showed a response rate of 75% and 58% for patients with AS in face/scalp and other primary sites (including five AS of the heart), respectively. The median time to progression (TTP) for the face/scalp group was 9.5 months, and for patients with AS at other sites was 7.0 months. 44 The ANGIOTAX study demonstrated clinical benefit of weekly paclitaxel for patients with metastatic or unresectable AS reporting a non-progression rate at 6 months of 24%, a median TTP and a median OS of 4 and 8 months, respectively. 45
Regarding targeted therapies, the most relevant clinical application in STS was observed in the treatment of gastrointestinal stromal tumors and dermatofibrosarcoma protuberans, revolutionizing the outcome of patients affected by these rare histotypes. For all other principal types of STS, the only approved tyrosine kinase inhibitor (TKI) is pazopanib, a multitargeted tyrosine kinase inhibitor, with activity against vascular endothelial growth factors 1, 2, and 3, and platelet-derived growth factors. Pazopanib demonstrated its activity in a randomized, placebo-controlled, phase III trial, showing a significant improvement in progression free survival (PFS) and a favorable trend in OS in patients with advanced non-adipocytic STS who had previously been treated with anthracycline- or ifosfamide-based chemotherapy. 46 However, it is not specified if any cardiac AS were included in the study.
There are few published data on pazopanib efficacy, specifically in vascular sarcomas. Considering the expression of pro-angiogenic growth factors, AS are inherently a target for antiangiogenic agents, thus the evaluation of vascular-targeted agents in these tumors is of particular interest. Recently, a retrospective study of patients with advanced vascular sarcomas, including AS, treated with pazopanib in real life practice was performed to specifically investigate its activity in these subtypes and provide a benchmark for clinical practice. 47 Results reported no significant difference in efficacy between cutaneous and non-cutaneous AS and radiation-associated and non-radiation-associated AS, showing an overall activity of pazopanib comparable with other STS subtypes. Once again, it is unclear if there were any cases of primary cardiac AS.
Based on the same assumptions, other monoclonal antibodies and TKI, such as bevacizumab, sorafenib and imatinib, mainly characterized by antiangiogenic mechanism of action, have been tested in AS alone or in combination with chemotherapy.48–52 In most trials, it is unclear whether primary cardiac AS are included within the study population, or, if they are, their number is too small to draw credible conclusions in the specific cardiac setting. The results of the principal studies are reported in Table 2. They were sometimes disappointing, sometimes interesting, but none eventually led to approval of these molecules in conventional treatment of AS.
Studies of monoclonal antibodies and tyrosine kinase inhibitors tested alone or in combination with chemotherapy in angiosarcoma.

AS, angiosarcoma; EHE, epithelioid hemangioendothelioma; mOS, median overall survival; mPFS, median progression free survival; pts, patients; RR, response rate; STS, soft tissue sarcoma.
A randomized phase III trial has recently compared the efficacy of pazopanib alone versus pazopanib plus TRC105 in patients with advanced AS. 53 TRC105 is a monoclonal antibody to endoglin, an essential angiogenic target highly expressed on tumor vessels that is distinct from VEGFR. Endoglin is also expressed directly on tumor cells in AS and is upregulated following VEGF inhibition. The rationale was that TRC105, targeting a pathway upregulated following VEGF inhibition, could complement and potentiate VEGFR TKIs, resulting in more effective angiogenesis inhibition and improved clinical efficacy when compared with pazopanib alone. An interim analysis following enrollment of 123 patients showed no meaningful efficacy for TRC105/pazopanib combination versus pazopanib alone in the face of greater toxicity especially concerning anemia, headache, and epistaxis. 54
Undifferentiated pleomorphic sarcoma
Undifferentiated pleomorphic sarcomas are cardiac sarcomas with no diagnostic histologic pattern or specific immunohistochemical profile. According to the most recent classification, undifferentiated pleomorphic sarcoma (UPS) replaces the now obsolete term pleomorphic malignant fibrous histiocytoma and includes intimal sarcoma (IS). 36 UPS generally arise in the left atrium, with a controversial prevalence depending on criteria adopted for diagnosis; in a broad definition, it represents the most common sarcoma type of the heart, accounting for at least 50% of cardiac sarcomas. 36
This classification of UPS will likely evolve in the coming years as more molecular and cytogenetic findings will be accumulated. Some authors have proposed intimal sarcoma as a distinct subtype of cardiac sarcoma, characterized by MDM2 amplification and overexpression. 20 However, this concept is not universally accepted, since the heart is lined by endocardium and not by intima, which are biologically distinct linings, though both derived by endothelium. In addition, MDM2 amplification is not a tumor-specific event, having been identified in other tumor types, in particular liposarcoma. As such, the term ‘undifferentiated pleomorphic sarcoma’ with or without MDM2 amplification is considered more appropriate, 55 as specified in the most recent WHO classification of tumors of the heart. 36
Globally, cardiac UPS have a complex genomic profile with alterations reported in all chromosomes. Whilst amplification of MDM2 is the most frequent event, other recurrent chromosomal imbalances have been detected, such as 4q12 amplification including KIT and PDGFRA, gain in 7p12 region including EGFR and loss of 9p21 region targeting CDKN2A.20,56 Moreover, TP53 alterations (mutations or protein accumulation) have been reported in 12–22% of UPS cases (all sites), some of which are in association with concurrent MDM2 amplification.57,58 To date, mutational analysis of only three cases of cardiac UPS have been performed.17–19 In one of these, a novel inherited loss of functional mutation of FH (p.E404D) and a recurrent somatic hotspot mutation of PIK3CA (p.H1047R) were detected. 17 In the other two cases, PDGFRB activating mutations (p.R709H and a p.E472D) have been identified, concurrent with PDGFRA amplification.18,19 These genetic changes are similar to those seen in IS of large vessels. In a cohort of 21 large arterial blood vessel IS, PDGFRA amplification was the most frequent abnormality (81% of cases), followed by MDM2 amplification (65% of cases) and EGFR amplification or aneuploidy (both accounting for 76% of cases). Interestingly, amplification of PDGFRA was found associated with receptor activation and, in some cases, concurrent activation of EGFR was detected. 59 PDGFRB mutations and kinase activation have been also identified in a small subgroup of IS. 59 Together, these data provide a rationale for investigating therapies that target PDGF receptors, EGFR, or MDM2 in cardiac UPS carrying these abnormalities. In particular, some studies have been conducted with imatinib, sorafenib and sunitinib reporting no objective response and median PFS and OS in the range of 1.9–2.5 months and 12.2–13.6 months, respectively.51,52,60
It is worth noting that other druggable mutations involving ALK, ATM/ATR and PTCH1 have been described in IS of the pulmonary artery, 61 which can occasionally extend to the heart. However, none of them were reported in primary cardiac UPS.
In brief, given the clonal heterogeneity of intimal sarcomas and the potential crosstalk between different signaling pathways, resistance to a single-agent tyrosine kinase inhibitor is a common event and durable disease control is very unlikely to be achieved. Simultaneous targeting of more than one relevant pathway is a theoretical approach that should be explored in prospective clinical trials, while paying attention to monitoring toxicity, especially in heavily pre-treated patients.
Other histologies
Several other histologic subtypes of cardiac sarcoma are recognized. However, each of them accounts for some 10% of cardiac sarcomas 36 and the number of tumors analyzed at molecular level reported in literature is extremely limited. For each subtype, we briefly describe the main molecular alterations identified in other tissues and, where available, in the cardiac setting.
Myxofibrosarcomas (MFS) are approximately 10% of sarcoma of the heart and thought to arise from the fibroblasts of the cardiac connective tissue. Very little is known about clinical presentations, pathologic features, treatments, outcome patterns and molecular landscape of MFS (Figure 2).

Local relapse of myxofibrosarcoma of the left atrium in a 74-year old woman. 18F-FDG PET/CT shows faint uptake (SUVmax = 4.8) in the left atrium (a). Cardiac MRI (b) and CT scan (c) confirm the voluminous, solid formation (47 × 24 mm) adhering to the lower wall of the left atrium with trans-mitral development.
Sun et al. recently published pooled analyses, collecting 31 cases of primary cardiac MFS. 62 Results revealed that they afflicted relatively young patients, with a mean age of 42 years. The most common cardiopulmonary symptom reported was dyspnea (64.3% of cases) and the most common location was the left atrium (58% of cases). The median survival time was 14 months, with major risk factors related to a poor prognosis represented by tumors ⩾40 mm in size or with high-grade. Regarding molecular information, some studies identified MFS as genetically highly complex and heterogeneous tumors, lacking specific or recurrent genetic fingerprints, with frequent gross amplifications and deletions. On the contrary, the abundance of point mutations is relatively low, with TP53 signaling and cell cycle checkpoint genes as more frequently altered pathways.63,64 We have recently reported a case of an elderly woman affected by MFS of the left atrium, describing its molecular features by whole transcriptome sequencing. 21 We confirmed a pattern of considerable complexity in the gene expression profile of MFS, which could justify the responsiveness to “classical” chemotherapy treatments (such as anthracyclines). Moreover, we found several genes co-expressed in MFS and AS, but not in IS, suggesting a putative similarity between MFS and AS, also in terms of response to gemcitabine, which is an active drug in this histological subtype. 65
Primary cardiac osteosarcomas account for approximately 10% of cardiac sarcomas. 36 Only a few cases in literature described their clinical course and some cytogenetic analysis data, reporting allelic losses or imbalances involving 1p36, 9p22, 10q23, 17q22, and 22q13. 66 In general, conventional high-grade osteosarcomas are tumors with complex karyotypes. They are characterized by chromosomal instability, with numerous gross genomic mutations and rearrangements. About 33% of cases have been reported to harbor chromothripsis events and one half of all osteosarcomas exhibit patterns of localized hypermutation that are termed kataegis. 67 Hundreds of genomic rearrangements have been identified in osteosarcomas, including recurrent rearrangements of TP53, RB1, MDM2 and CDKN2A gene fusions. 68 Up to 80% of tumors carry TP53 biallelic inactivation, predominantly through structural variations in intron 1. On the contrary, low rates of single nucleotide variations (mainly involving TP53 and RB1) are reported. 67
Aggressive surgery and postoperative polychemotherapy (based on doxorubicin, cisplatin, ifosfamide, etoposide, mitomycin, methotrexate, and/or gemcitabine) are the most important strategies adopted in cardiac osteosarcoma treatment. However, the prognosis of this tumor is still very poor. 69
Rhabdomyosarcomas (RMS) are tumors showing skeletal muscle differentiation and they account for only 0–5% of all cardiac sarcoma. Most cases appear in children at a mean age of approximately 14 years, and the histologic features are usually of the embryonal subtype. 36
Molecular analysis of only one case of cardiac RMS, harboring a KRAS mutation (codon 13 G>A transition), has been reported. 15 Similarly, alteration of RAS pathway (H/K/N-RAS) has been detected in extra-cardiac RMS, together with mutations of TP53, PIK3CA, CTNNB1, FGFR4, BCOR and FBXW7, and loss of heterozygosity at 11p15.5. 70 In this sense, precision medicine strategies for treating RMS could be developed. New potent selective dual RAF/MEK inhibitors are under investigation and preliminary studies have demonstrated effect on RMS cell lines and on mice. 71
Leiomyosarcomas (LMS) account for less than 10% of cardiac sarcoma. Histologically, they are composed of elongated and closely spaced spindle cells with smooth muscle differentiation. Wang et al. reviewed clinical presentations, imaging, pathology, treatments, and outcomes of 79 patients affected by primary cardiac LMS. 72 The mean age at onset was 48 years and the most frequent complaint at diagnosis was symptom of obstruction. Primary cardiac LMS tend to be biologically more aggressive compared with their counterparts in other sites with 5-year overall survival and recurrence-free survival rates of 25% and 15%, respectively. Complete resection, radiotherapy, and conventional chemotherapy/target therapy (anthracyclines, gemcitabine/docetaxel, gemcitabine/dacarbazine, trabectedin, pazopanib) represent the only treatment options. Expanded molecular profiling of cardiac LMS could potentially reveal actionable targets for personalized treatment strategies helping to increase the life expectancy. Currently, little is known about its molecular heterogeneity. Generally, defects in DNA repair and chromosomal maintenance (including mutations of TP53, ATM, RB1, and ATRX) are central to the biology of LMS. 73 However, concerning cardiac LMS, only a case harboring a HRAS mutation has been reported. 22
Synovial sarcomas (SS) can occur anywhere and in any age group, but frequently manifests in the distal extremities of young adults. Primary cardiac SS is an exceptionally rare entity, accounting for approximately 5% of cardiac sarcomas involving either the pericardium or chambers, with a striking male predominance. They can be either monophasic (more common; spindle cells only) or biphasic (less common; epithelial and spindle cells). From the molecular point of view, SS have a relatively stable genome and they are characterized by the pathognomonic reciprocal chromosomal translocation t(X;18) (p11.2;q11.2), which results in the fusion of the SS18 (formerly SYT) gene on chromosome 18 with one of the SSX genes on chromosome X (SSX1, SSX2, or rarely SSX4). 74 It was demonstrated that SS18-SSX fusion associates with SWI/SNF and Polycomb chromatin complexes to dysregulate gene expression through the disruption of epigenetic regulation. 75 This translocation, detected by reverse transcription-polymerase chain reaction, fluorescence in situ hybridization, or next generation sequencing, is considered to be a specific cytogenetic abnormality diagnostic of SS (all sites). Other than the SS18-SSX fusion gene, no other aberration has been reported in cardiac SS.23–35
Current multimodal treatment of cardiac SS includes surgical resection, radiation, chemotherapy and heart transplantation, the latter for selected cases (i.e. young patients with lower grade, small size, and less aggressive tumors). In general, numerous studies show that SS tend to have better survival rates and higher chemosensitivity than other STS subtypes, but these results mainly refer to SS of the extremities. Standard first and subsequent-line of chemotherapy for advanced, recurrent, and metastatic SS are represented by doxorubicin and ifosfamide, high-dose ifosfamide, and trabectedin. Investigational trials exploring multi-TKI agents (imatinib, pazopanib, regorafenib) have been conducted with mixed results. More recently, there has been some progress toward immunotherapeutic strategies. In particular, NY-ESO-1 is a cancer testis antigen, which is expressed in a substantial percentage of SS (49–76%). In this sense, treatment of patients with NY-ESO-1-positive tumors with genetically engineered lymphocytes seems promising and some clinical trials are currently ongoing.76–79
In conclusion, several recurrently altered genes have been identified in cardiac sarcomas mainly in AS and UPS, while adequate molecular characterization of rarer histologies is still lacking. Ideally, deeper and wider genetic analysis should be performed to complete our knowledge of the molecular landscape of all type of sarcoma of the heart.
Future perspective
Primary sarcomas of the heart are rare neoplasms. With manifold clinical manifestation, critical location and scarce evidence from literature, diagnosis of these tumors is often belated, clinical management significantly complex and outcome very poor. Therefore, there is an urgent need of more adequate tools for early diagnosis and more effective treatment. The molecular characterization of heart tumors could unfold potentially novel, druggable targets. Most literature on cardiac tumors consist of case reports and few articles provide details on their molecular profile. Even if several recurrently altered genes have been identified in cardiac AS and UPS, deeper and wider analysis is needed to complete the genetic footprints of these tumors and other cardiac sarcomas (Figure 3a). Most of these genes induce proliferation and angiogenesis through the activation of PI3K/AKT, PLCG1/PKC, and RAS pathways. These findings theoretically support the use of several targeted therapies, including TKI or monoclonal antibodies against KDR/EGFR/PDGFR and small molecules against their downstream signaling effectors (Figure 3b). Simultaneous targeting of more than one relevant pathway should be explored.
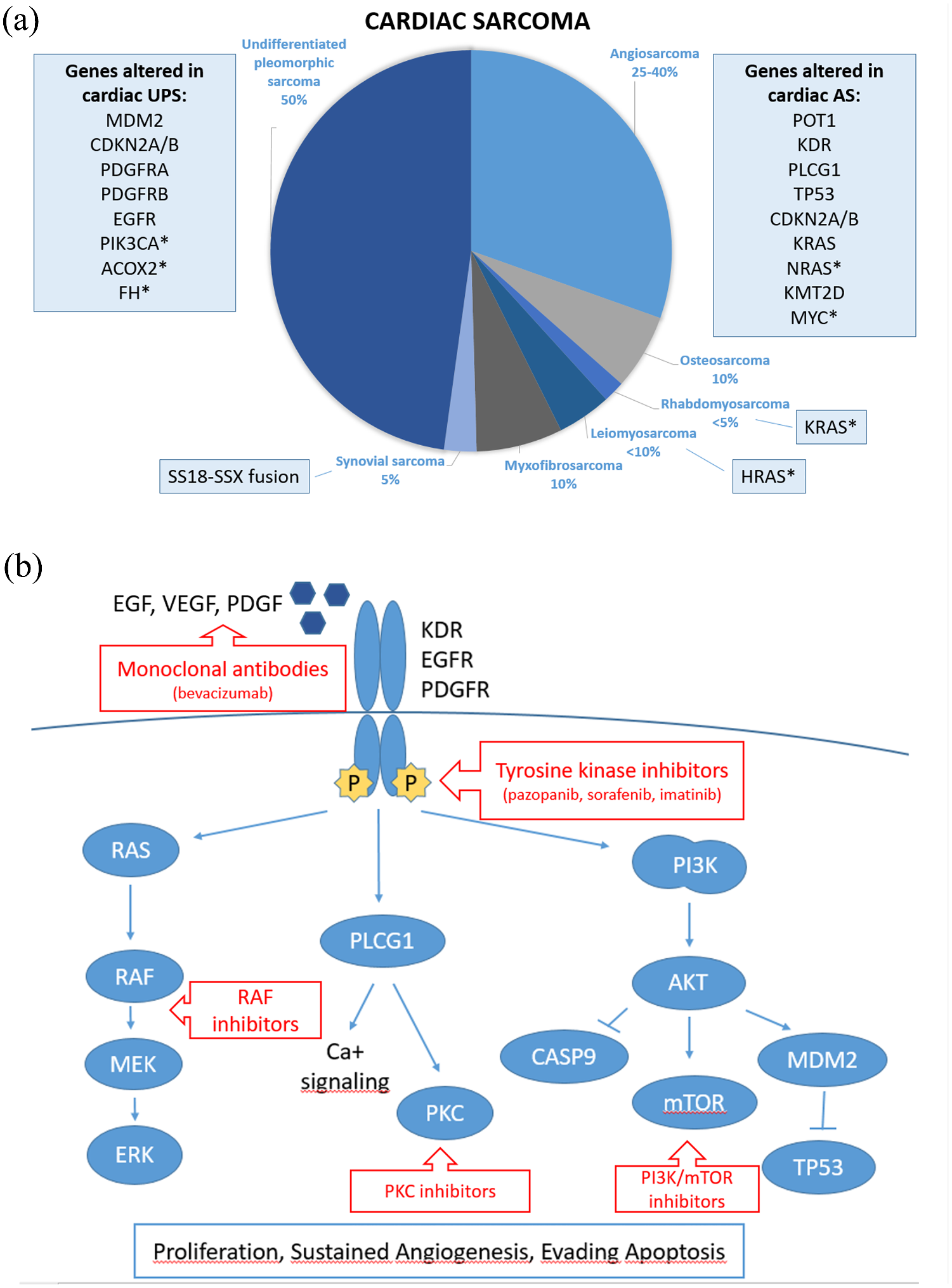
Mutational landscape and potential targeted therapy in cardiac sarcoma. (a) Frequency of each cardiac sarcoma histotype and altered genes identified so far. *indicates mutation reported in a single case. (b) Signaling pathways altered in primary cardiac sarcoma and potential therapeutic targets.
Next generation sequencing has revolutionized the study of genomics and, with decreasing cost per sample and wider accessibility of sequencing platforms, it is likely that more cardiac sarcomas will be characterized in the next future. However, molecular characterization without functional studies is insufficient for the evaluation of novel therapeutic strategies in this setting. The use of tumor models for functional evaluation of alterations identified in cardiac sarcoma is therefore essential and will require the development of adequate in vitro and in vivo models. In this context, induced pluripotent stem cells or patient derived xenograft models could be used, allowing a better comprehension of cardiac sarcoma’s biology. The hope would be to prospectively characterize and classify tumors by molecular behavior such that subtyping would be based on signal transduction pathway defects, ultimately helping to enrich the therapeutic weapons against this aggressive tumor.