
Abstract
We report the case of a woman nearing 70 years old who was admitted to the hospital with a complaint of “epigastric distension for 1 month”. Her main signs and symptoms were progressive abdominal distension and occasional abdominal pain. Computed tomography suggested an abdominal mass. She had a surgical history of synovial sarcoma (SS) of the lungs. After admission, she was diagnosed with jejunal SS following a puncture biopsy and laparoscopic surgery. This disease usually occurs in the soft tissues of the limbs, and it is extremely rare for SS to originate in the jejunum. The morphologic heterogeneity of SS overlaps with other tumors and makes the diagnosis particularly difficult. Imaging studies usually lack specificity; however, measuring multiple immunohistochemical markers can greatly assist in the diagnosis and differential diagnosis of SS. This case not only enriches our understanding of SS and describes a rare site of origin, but also emphasizes the importance and challenges of achieving an accurate diagnosis. Immunohistochemical and molecular biological testing have important roles in the definitive diagnosis, highlighting the need for precise and innovative diagnostic and therapeutic approaches in SS.
Keywords
Introduction
Synovial sarcoma (SS) is a rare malignant mesenchymal tumor exhibiting partial epithelial differentiation, and accounts for 5% to 10% of all soft tissue sarcomas. 1 SS occurs predominantly in the soft tissues of the extremities, especially the lower limbs, and develops rarely in joints or bursae, with less than 5% of cases originating in these structures. 2 SS has been documented in almost every anatomical site in addition to the common extremity sites, but is rarely seen in the skin, abdomen, and pelvis. 2 Histologically, SS can be categorized into biphasic, monophasic, and poorly differentiated types. 3 SS presents with a complex and diverse morphology that overlaps markedly with other soft tissue tumors, which often leads to misdiagnosis or missed diagnoses. 1 Notably, our institution recently treated a patient presenting with heterochronous multiple primary SS occurrences. SS developed separately in both the lungs and jejunum within 2 years, which is an uncommon scenario. We report the details of this case, including the clinicopathological characteristics; thereby, enhancing the understanding and recognition of SS in rare locations, to improve diagnostic accuracy.
Case presentation
A woman nearing 70 years of age was admitted to our hospital with a 1-month history of “episodic epigastric distension and pain”. She presented with no nausea, vomiting, chills, fever, melena, or hematochezia, and her bowel movements and intestinal gas passage were regular. Initial computed tomography (CT) at a local hospital indicated a space-occupying lesion in the abdominopelvic cavity and thrombosis in the superior mesenteric vein. The patient was admitted to the Interventional Radiology Department for further investigation and treatment. She had a history of hypertension and diabetes for 10 years, which were well-controlled with oral medications. Two years earlier, after being admitted to another institution with a complaint of >2 months of lower-limb edema, a lung tumor was discovered, and she underwent left lower lobectomy. Postoperative pathology revealed a left lower lung SS measuring 5.0 cm × 4.0 cm × 3.0 cm, with vascular invasion, no nerve involvement, and negative bronchial margins. No lymph node metastasis was detected (0 of 4 nodes). Immunohistochemistry revealed the following: vimentin (+), cluster of differentiation (CD)99 (+), B-cell lymphoma-2 (Bcl-2) (+), cytokeratin (CK): partially positive, epithelial membrane antigen (EMA): partially positive, CK7: partially positive, CD56 (±), actin (−), desmin (−), smooth muscle actin (SMA) (−), CD34 (−), podoplanin (D2-40) (−), CD68 (−), lysozyme (−), chromogranin A (CgA) (−), synaptophysin (Syn) (−), glial fibrillary acidic protein (GFAP) (−), S-100 (−), carcinoembryonic antigen (CEA) (−), transcriptional intermediary factor‐1 (TIF-1) (−), programmed death-ligand 1 (PD-L1) (−), PD-1 (−), and Ki-67 (>10%). The patient received no postoperative treatment and was not followed-up for 2 years.
Clinical examination findings on admission were as follows: body temperature: 36.4°C, respiratory rate: 20 breaths/minute, pulse rate: 97 beats/min, blood pressure (BP): 112/62 mmHg, and body mass index (BMI): 29 kg/m2. The patient was generally in good condition, moving freely, and she responded appropriately to any questions. There were no observable abnormalities in the joints of the extremities, and cardiovascular and pulmonary examination findings were unremarkable. Her abdomen was distended, with a palpable moderately mobile mass measuring approximately 15 × 15 cm in size located in the epigastric region. No signs of rebound tenderness or muscular tension were present across the abdomen, and bowel sounds were normal. During the first year after resection of the lung tumor, she began to notice mild abdominal distension and occasional pain, but did not pursue a medical assessment. It was not until the second postoperative year, as the symptoms of abdominal distension increased, that she came to our hospital and was diagnosed with an abdominal space-occupying lesion.
The preliminary diagnosis was as follows: huge abdominopelvic tumor, superior mesenteric vein thrombosis, primary hypertension, and type 2 diabetes. Abdominal CT revealed a mass in the right abdominopelvic region, suspected to be a stromal tumor, measuring approximately 130 mm ×120 mm (Figure 1). The mass had a heterogeneous density and irregular enhancement on contrast-enhanced images. The boundary between the mass and the adjacent intestines was unclear, and thickening of the surrounding peritoneum and mesentery was observed. Thrombosis was evident in the superior mesenteric vein and accompanied by significant ascites in the abdominopelvic cavity.

Abdominal enhanced CT showing a mass with uneven density and uneven edge enhancement. CT, computed tomography.
Chest CT revealed scattered small nodules in both lungs (Lung Reporting and Data System (LU-RADS) category 2). The electrocardiogram (ECG) was normal, and routine blood and biochemical test results were unremarkable. The tumor markers, carbohydrate antigen (CA)125 and human chorionic gonadotropin (hCG) were slightly elevated, whereas CEA levels were within normal limits. Biopsy suggested a spindle cell tumor and a malignant mesenchymal tumor. After consultation, the patient underwent surgery in our department, which comprised laparoscopic resection of the massive abdominopelvic tumor and segmental small intestinal resection and anastomosis. Intraoperatively, approximately 5000 mL of pale-yellow ascitic fluid was found. No distant metastases were observed in the liver, spleen, or peritoneum. The tumor measured approximately 15 × 15 × 15 cm and had an irregular lobulated appearance with a complete capsule. Satellite lesions were visible around the main lesion. The tumor was cystic and solid, and, internally, contained soft, loose tumor tissue. The tumor infiltrated and adhered to the small intestine and mesentery, and originated in the jejunum 120 cm from the ligament of Treitz. A tumor thrombus was observed in the basal vein, causing significant mesenteric edema. The main blood supply originated from the superior mesenteric vessels of the upper jejunum. The tumor was completely excised, with an estimated blood loss of approximately 50 mL (Figure 2).

Successful and complete removal of the tumor. (a) The tumor originated from the jejunum and grew irregularly. Satellite foci are visible. (b) Intraoperative confirmation that the tumor originated from the jejunum and (c) completely resected tumor.
Postoperative pathology revealed a small intestinal SS, World Health Organization (WHO) grade 3. The tumor affected the mucosal, submucosal, and serosal layers and mesentery of the small intestine. Hematoxylin and eosin (HE) staining revealed biphasic differentiation (Figure 3a). Immunohistochemistry (Figure 3b–d) revealed the following: vimentin (+++), CK7 (++), EMA (scattered +), CD117 (−), discovered on gastrointestinal stromal tumor-1 (DOG-1 (−), succinate dehydrogenase (SDH) (+), CD34 (−), CD31 (−), E-26 transformation-specific-related gene (ERG) (−), transcription factor E3 (TFE3) (−), signal transducer and activator of transcription 6 (STAT6) (−), trimethylation at lysine 27 of histone H3 (H3K27Me3) (+), integrase interactor 1 (INI-1) (+), Wilms’ tumor 1 (WT1) (−), S-100 (–), SMA (−), desmin (−), calretinin (focal +), and Ki-67 (>50%). Fluorescence in situ hybridization (FISH) evaluation was performed, and the results suggested the presence of specific SS18 gene rearrangement (separation of red and green signals) confirming the diagnosis of SS (Figure 4).

HE staining and immunohistochemistry of the postoperative specimens. (a) HE staining (×200): biphasic tumor composed of spindle cells and epithelioid cells. (b) Immunohistochemistry (×200) showing vimentin (+++) cells. (c) Immunohistochemistry (×200) showing CK7 (++) cells and (d) immunohistochemistry (×200) showing EMA (scattered +) cells. HE, hematoxylin and eosin; CK, cytokeratin; EMA, epithelial membrane antigen.
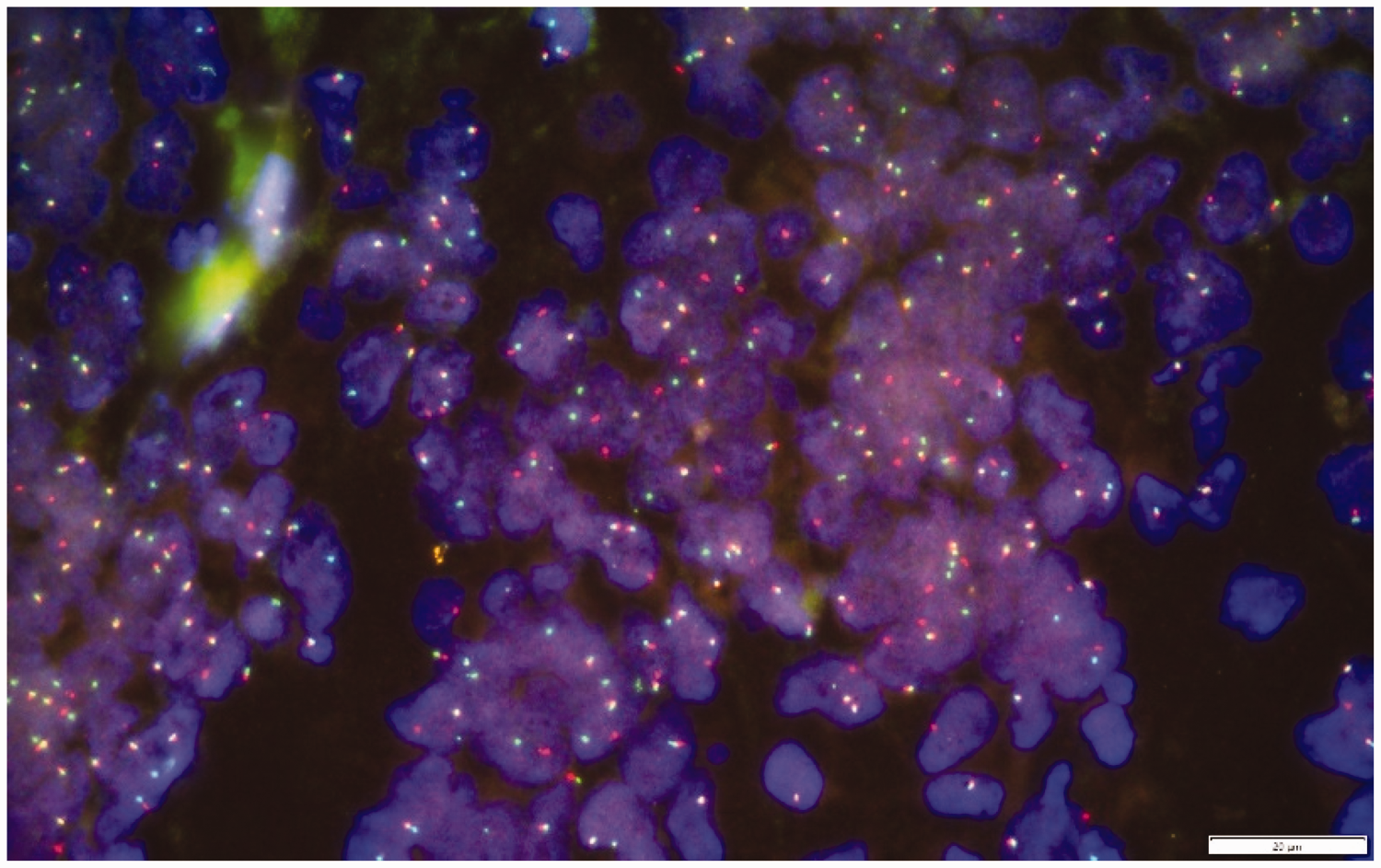
FISH test results indicating SS18 gene rearrangement. FISH, fluorescence in situ hybridization.
The patient was safely discharged 1 week postoperatively. No significant intraoperative or postoperative complications occurred. After discharge, she was advised to undergo chemotherapy at the oncology hospital, but failed to undergo regular evaluations as her overall condition continued to deteriorate, with poor mental health and weight loss. Physicians at the oncology hospital assessed the seriousness of her condition and the development of multiple organ failure and did not consider chemotherapy an option. Owing to the lack of regular follow-up, it was difficult to obtain radiological evidence of metastasis. The patient died of multiple organ failure 4 months after discharge.
This study was performed in accordance with the principles of the Declaration of Helsinki and approved by the Ethics Committee of the Affiliated Hospital of Zunyi Medical University (KLL-2023-610), in March, 2023. Because we have de-identified all patient details, written informed consent from the patient or her family was not required for publication of this case report. This case report was written in accordance with the CARE guidelines. 4
Discussion
SS originates from the mesenchymal tissues, is highly malignant, and predominantly manifests near the knee joint; although SS can develop in other regions, such occurrences are relatively rare. 5 The head and neck region, abdominal wall, retroperitoneum, mediastinum, pleura, lungs, and other organs are less common locations; 6 the digestive tract is an infrequent site of SS manifestation. 7 SS originating primarily in the jejunum is rare. Wang et al. 8 performed a literature review using the PubMed database and identified 15 reported cases of SS in the lower digestive tract. Among these, only one SS was found in the jejunum, and it was the monophasic type. The concurrent multiple primary occurrences of SS in both the lungs and jejunum observed in our patient is the first such reported case, to our knowledge. At the time of the initial diagnosis of SS in the patient’s left lower lung, no SS or other tumors were found in the intestine or elsewhere. Notably, jejunal SS was found 2 years after complete resection of the SS in the left lower lung. This long time interval and because the tumors were found in different sites (lung and jejunum) strongly support the idea that the two lesions were independent heterochronous multiple primary SS. Although we could not rule out the possibility that the jejunal SS was not detected at the time of the initial diagnosis because it was too small, considering the clinical presentation and tumor onset in different sites, we are inclined to believe that the two lesions were independent primary tumors.
A characteristic of SS is mesenchymal differentiation coupled with an epithelioid character. 2 Consequently, histological classification primarily encompasses three types: monophasic, biphasic, and poorly differentiated. 3 This diverse differentiation leads to various morphologies, making radiological diagnosis challenging. Imaging examinations, including CT and magnetic resonance imaging (MRI), do not display typical specificities or consistent patterns. 9 Multiple findings have been identified for SS manifestations on MRI that can predict high-grade lesions, including the absence of calcifications, presence of hemorrhage, and triple signal intensity. 10 However, not all SSs exhibit the typical “triple sign”. Sedaghat et al. 11 performed MRI in 15 histologically-confirmed primary SS cases, revealing that 60% of the tumors displayed the triple sign. Liang et al. 9 identified the triple sign in only 50% of their cases, while Ashikyan et al. 12 noted its presence in 80% of SSs. Contrast-enhanced CT can be performed when MRI is contraindicated or unavailable. Compared with MRI, CT provides better visualization of soft tissue calcification and local bone reactions. Compared with heterogeneous muscles in large lesions, SS has a lower signal. 13 With SS, CT typically shows a non-infiltrating mass with heterogeneous attenuation, similar to or slightly lower than muscle attenuation, frequently accompanied by peripheral punctate calcifications. 6 Reportedly, 89% to 100% of cases exhibit heterogeneous post-contrast enhancement, which aids in differentiating SS that initially manifests as cystic lesions or hematomas. 6 Given the rarity of SS in non-limb locations, a definitive diagnosis requires consideration of the lesion site. Differential diagnosis of other tumor types, such as angiosarcoma and gastrointestinal stromal tumors remains a challenge. In our case, CT revealed a tumor with uneven marginal enhancement, with the internal portion showing no enhancement. Imaging indicated only a close relationship with the intestinal wall, leading to a radiological diagnosis that tended toward gastrointestinal stromal tumors. Given the absence of joint symptoms and the primary location of the tumor within the abdominal cavity, SS is not typically considered among abdominal tumors. The diagnosis primarily relies on pathological examinations beyond imaging studies.
The diagnosis of SS currently involves a comprehensive approach comprising clinical manifestations, histological morphology, and immunohistochemistry, and pathological examination remains the gold standard for definitive diagnosis. Owing to the overlap of SS with other soft tissue sarcomas with respect to the clinical features, histological morphology, and immunophenotype, precise diagnosis of SS presents certain difficulties. Various tumor markers in SS have limited diagnostic value owing to their lack of sensitivity and/or specificity, and the most helpful markers comprise EMA, CK7, CK19, vimentin, calponin, Bcl-2, CD99, human transducin-like enhancer of split 1 (TLE1), and other proteins.14,15 A series of immunohistochemical markers have been proposed to support the diagnosis of SS, but no single marker or combination of markers can definitively confirm or exclude the diagnosis of SS. 6 Although some markers overlap with those of other soft tissue sarcomas in SS, leading to misdiagnosis, the combined detection of multiple markers has important implications in the differential diagnosis of SS. SS has characteristic immunohistochemical features, such as positive staining for AE1/AE3, CK7, and EMA, but negative staining for CD117, CD34, desmin, and S100 protein. 16 The most sensitive epithelial differentiation marker is EMA, with over 90% of SSs showing focal positive expression. 17 Additionally, diffuse wavy proteins and Bcl-2 positivity are visible in most SSs, 18 and CK7- and CK19-positivity appear to be limited to SS only. 2 Immunohistochemistry revealed strong and diffuse nuclear staining for the transcriptional repressor TLE1 in most SS samples. 19 New York esophageal squamous cell carcinoma-1 (NY-ESO-1) is also strongly expressed in most SSs, aiding in distinguishing SS from other spindle cell tumors. 20 In our case, epithelial markers, such as CK and EMA, showed varying degrees of positivity; the mesenchymal tissue marker, vimentin, was strongly positive; desmin and SMA were negative, as was the neuroendocrine and neurilemmoma differentiation marker, S-100. All CD family markers were negative, and the cell cycle regulatory protein markers, Bcl-2 and Ki-67 were positive. Notably, the diagnosis in our case relied on the positive expression of epithelial markers, mesenchymal markers, and cell cycle regulatory protein markers, combined with the coexistence of nested and spindle cells with HE staining. Previous reports of the immunohistochemical findings in cases of rare sites of SS, such as in our case, indicate that CK-, EMA-, and vimentin-positivity is common. Therefore, positive testing for these three markers can be considered the most practical and sensitive basic combination in immune marker testing for the diagnosis of SS.
Symptoms in patients with SS are non-specific; painless swelling is the most common presentation. 6 However, various symptoms may be associated with different sites and tumor sizes, such as swelling or pain caused by compression of adjacent tissues. Furthermore, patients with intra-abdominal SS may present with abdominal pain, a feeling of bloating, and dyspepsia, 21 as in our case, in which the patient presented primarily with epigastric distension. SS should first be distinguished from gastrointestinal stromal tumors (GIST). 8 GISTs are typically composed of spindle-shaped and epithelioid cells, whereas biphasic SS is often composed of spindle cells and glandular epithelial components. 8 For most GIST cases, positive staining for CD117, CD34, and DOG1 combined with negative staining for epithelial markers confirms the diagnosis. 8 Given these rare exceptions, it is best to supplement molecular testing with identification of mutations in KIT and platelet-derived growth factor receptor alpha (PDGFRA) to further confirm a GIST diagnosis. 8 There is marked variability in the expression of various markers in SS, as detected by immunohistochemistry, which presents certain challenges in diagnosis, necessitating the identification of a reliable diagnostic method. With advancements in molecular biology, a highly specific SS18-SSX (also known as SYT-SSX) fusion factor has been identified in SS. 6 The chromosomal translocation t(x;18)(p11.2;q11.2) results in the fusion of the SSX gene (either SSX1, SSX2, or SSX4) on the X chromosome with the SS18 gene on chromosome 18. 22 Studies have shown that this can be detected in approximately 95% of patients with SS. 20 The detection of t(X;18) using cytogenetic methods, such as FISH, reverse transcription polymerase chain reaction (RT-PCR), or next-generation sequencing (NGS) is considered the gold standard for diagnosing SS, 23 highlighting the importance of these techniques in clinical diagnosis.
SS is a high-grade invasive sarcoma with a challenging treatment landscape and 5- and 10-year survival rates of approximately 36% and between 10% and 30%, respectively. 24 The prognosis is worse in patients diagnosed with metastases, with a 3-year survival rate of only 27.2%. 25 Most researchers suggest surgical resection as the primary intervention, asserting its efficacy when complemented by radiotherapy and chemotherapy. 26 However, for patients with metastatic or unresectable SS, palliative chemotherapy is used, 5 with doxorubicin as the standard first-line drug. The combination of ifosfamide and doxorubicin provides even more benefit. 27 There are currently limited therapeutic drugs available for targeted therapy, and pazopanib remands the only targeted drug approved for the treatment of SS. 27 The choice of a specific treatment for patients with SS is based primarily on the stage of the disease and the patient’s specific situation. Neoadjuvant chemotherapy is not commonly used but is considered induction therapy in certain circumstances to improve surgical outcomes for high-risk sarcomas of the extremities and chest wall. 28 Another study reported that for children and adolescents with intermediate- or high-risk tumors (i.e., >5 cm, lymph node involvement, positive resection margins), adjuvant or neoadjuvant chemotherapy is usually used, and the most common drugs are ifosfamide and doxorubicin. 20 Although the role of (neo) adjuvant chemotherapy in adult patients with SS is controversial, its demonstrated survival enhancement in pediatric SS patients makes its use in adult patients at high risk of recurrence an option to consider. 5 In the treatment of advanced disease, anthracyclines, isocyclophosphamide, trimethoprim sodium, and pazopanib have been chosen on the basis of their proven effectiveness in inhibiting tumor growth. 29 Nonetheless, there are limitations to these treatments, such as adverse events associated with chemotherapy and variable individual responses to treatment. Currently, a deeper understanding of the unique biology of SS is driving research into new therapies. For example, immunotherapeutic strategies targeting NY-ESO-1 have demonstrated potential efficacy, 30 but further clinical trials are needed. Similarly, metabolic therapies targeting argininosuccinate synthetase (ASS1) enzyme defects are an emerging research focus. 31 Future research must focus on developing more effective therapeutic approaches, especially given the unique biology of SS. Ongoing clinical trials, such as immunotherapy targeting NY-ESO-1 and metabolic therapies targeting ASS1 enzyme defects, may provide new strategies to improve outcomes for SS patients. 29 Therefore, it is critical to enroll suitable patients in these clinical trials. Overall, although current therapeutic approaches for SS have been effective, the limitations of these approaches, and future research directions, suggest that a deeper understanding of the biology of SS and the development of new therapeutic strategies are essential to improve patient survival and quality of life.
Conclusion
In this case, we emphasized the value of a combination of diagnostic tools, including highly-specific immunohistochemical markers and molecular biology techniques, particularly the detection of the t(x;18) (p11.2;q11.2) chromosomal translocation. Although imaging can provide an initial guide, diagnostic conclusiveness relies heavily on detailed pathologic examination, which in this case, was supported by the use of FISH to detect the t(X;18) translocation, a key marker in the diagnosis of SS. In the future, the search for more economical and reliable diagnostic methods will be a focus. In the meantime, this case reminds us that vigilance and an open diagnostic perspective are necessary for rare sites of SS onset.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605241233953 - Supplemental material for Synovial sarcoma of the viscera (lung and jejunum): a case report
Supplemental material, sj-pdf-1-imr-10.1177_03000605241233953 for Synovial sarcoma of the viscera (lung and jejunum): a case report by Jixin He, Jiwei Wang, Lina Yang, Kai Wang, Maijian Wang and Jianguo Li in Journal of International Medical Research
Footnotes
Acknowledgements
The authors are grateful for the patient’s consent to be part of this study.
Author contributions
JH, MW, and JL managed the patient’s care. JH, JW, and LY contributed to data collection and analysis. MW and KW contributed to the radiology and pathology analyses. JH drafted the manuscript. MW and JL revised the manuscript for intellectual content. All authors critically revised the manuscript and agree to be fully accountable for the integrity and accuracy of the work. All authors have read and approved the final manuscript.
Data availability statement
Relevant data for this study are available from the corresponding author upon reasonable request.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Funding
The authors disclose receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Regional Project of the National Natural Science Foundation of China [grant number 81969105], the Training Plan for Clinical Famous Doctors of Zunyi Medical University [grant number RC220230405], and the Excellent Youth Talent Training Program of ZMU affiliated hospital [grant number RC220220904].
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.