
Abstract
Improvements in median overall survival in the advanced oesophagogastric (OG) setting have plateaued, underlining the need for improved therapeutic approaches in this patient population. Immunotherapeutics are inducing unexpected durable responses in an expanding list of advanced disease indications. Although OG cancers have traditionally been considered to be more challenging to treat with immunotherapy than some other malignancies because of their variable tumour mutational burden and relative scarcity of infiltrating T cells, immune checkpoint inhibitor (ICPI) trials conducted over the last few years suggest there is an important role for these treatments. ICPI efficacy may be demonstrated in specific molecular subtypes of OG cancer. This review outlines the improvements in defining predictive biomarkers of responsiveness to ICPIs. Increasingly, identification of an expanding list of ICPI resistance mechanisms will drive biomarker-directed research. In addition, the specific rationale to combine ICPIs with chemotherapies, radiotherapies, targeted therapies and other novel immunotherapeutic drugs will be discussed.
Keywords
Introduction
Current stagnation in survival improvements in treatments of advanced oesophagogastric cancers
Gastric cancer is the fifth most common cancer, and the third leading cause of cancer-related deaths worldwide. 1 Oesophageal cancer is less common, however it accounts for a disproportionate amount of the total cancer deaths each year internationally. 1 Most cases of both cancers are diagnosed at a late stage, when treatment options are usually restricted to palliative chemotherapy. Although serial incremental survival benefits particularly in gastric cancer have been observed, 2 for most populations with advanced disease median overall survival (mOS) has plateaued. This is despite the enhanced efficacy added by targeted therapies, including trastuzumab in human epidermal growth factor receptor 2 (HER2)-positive disease in the first-line 3 and anti-vascular endothelial growth factor receptor 2 (VEGFR2) antibody ramucirumab in the second-line setting.4,5 Thus, there is a clear unmet clinical need for better therapies in advanced oesophagogastric (OG) cancer. This review will focus on some of the immunotherapeutic approaches being developed, with a major emphasis on adenocarcinoma of the stomach and oesophagus.
Current position of immunotherapies in OG cancers
Across both haematological and solid tumour indications, the expansion of the use of immunotherapy and specifically immune checkpoint inhibitors (ICPIs) is dramatically shifting one of the treatment goals in a proportion of patients with advanced disease: for some, maintained durable responses with elevation in the tail of overall survival (OS) curves has become a conceivable treatment goal. The immune landscape in OG cancer is, however, thought to differ considerably from other types of cancer, including melanoma and non-small cell lung cancer (NSCLC), in which ICPI therapies were first licensed. These differences include a mid-to-high tumour mutational burden (TMB) range 6 and variable densities of infiltrating proinflammatory T cells. 7 Thus, OG cancers may be predicted to be less likely to respond to a shift towards the enhanced immunogenicity invoked by ICPIs seen in tumours with the highest TMB. However, a closer look at the biology of immune checkpoint molecules may help define a specific role for their targeting in OG cancers. The number of identified immune checkpoint molecules is expanding but include cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed death 1 (PD-1) and its known ligands PD-L1 and PD-L2.
CTLA-4 is an inhibitory receptor constitutively expressed on regulatory T cells and activated cytotoxic T cells. It is a CD28 homologue with much higher binding affinity for the costimulatory immune-activating T-cell receptor, B7. 8 Anti-CTLA-4 antibodies induce an antitumour response, probably by inducing an immune priming phase and reducing regulatory T-cell-mediated suppression of inflammatory responses. 9 Some of the immune-mediated toxicities described for this class of therapeutics are likely due to their potential to ablate the role of regulatory T cells in maintaining peripheral tolerance. 8 PD-1 is a negative costimulatory receptor expressed mainly on activated T cells 10 which downregulates excessive immune responses by binding to its ligands, PD-L1 and PD-L2. 11 PD-L1 is constitutively expressed in various tissues and on an expanding list of several tumour types, including OG cancer.12,13 Specifically, PD-L1 expression has been detected in more than 40% of human gastric cancer samples in several studies, 12 though its overexpression in oesophageal cancer is estimated to be considerably lower at around 15–20%. 13 PD-1 blockade is postulated to work during the T-cell effector phase to restore the immune function of exhausted T cells following extended or high levels of antigen exposure, as occurs in advanced cancer. 8 Figure 1 highlights the roles these immune checkpoint molecules have on cancer regulation, and suggests that their inhibition may have different mechanisms of antitumour efficacy.

CTLA-4 and PD-1 pathway inhibition. CTLA-4 inhibition allows for activation and proliferation of more T-cell clones, and reduces Treg-mediated immunosuppression. PD-1 pathway inhibition restores the activity of antitumour T cells that have become quiescent.
Two anti-CTLA-4 murine antibodies, ipilimumab and tremelimumab, have been assessed in OG cancer. A phase II study of tremilumumab in the second-line treatment of unselected advanced OG cancer showed an overall response rate (ORR) of 5% with a mOS of 4.8 months. 14 Of note, and supportive of the proposed mechanism of action of ICPIs, the sole responder continued treatment beyond 2 years. A phase II study assessed the efficacy of ipilimumab as maintenance treatment immediately after first-line chemotherapy in locally advanced irresectable or metastatic gastroesophageal cancer compared with best supportive care. 15 No difference was demonstrated in mOS between the two study arms.
After these less than encouraging results, enthusiasm for the potential benefits of ICPIs in OG cancer was reignited by the preliminary results demonstrated in the pembrolizumab, KEYNOTE-012 study. 16 Pembrolizumab is a humanised monoclonal antibody designed to bind to PD-1 and thus block the interaction between PD-1 and its ligands. In this study, patients with PD-L1-positive disease (>1%) were enrolled for pembrolizumab as first- or subsequent-line therapy (n = 39). An ORR of 22% with median duration of responses of 40 weeks was observed. The 6-month progression-free survival (PFS) rate was 26%, and the mOS was 11.4 months, with a 12-month OS rate of 42%. Of note, over 50% of patients had received three or more lines of therapy. Pembrolizumab has subsequently been licensed by the US Food and Drug Administration (FDA) in third-line or more advanced PD-L1-positive (>1%) gastric cancer.
Nivolumab is a humanised monoclonal antibody to PD-1, which has been evaluated in a number of studies in gastric cancer. Most recently, the pivotal phase III, placebo-controlled, randomised and licensing study was reported for nivolumab in third- or subsequent-line therapy. The ONO12 (ATTRACTION-2) study recruited in Korea, Japan and China only, and thus consisted entirely of Asian patients. 17 Patients were not selected according to PD-L1 status. In this large study (n = 493), patients were randomised in a 2:1 fashion to nivolumab or placebo. Nivolumab resulted in statistically superior OS, PFS and ORR (11.2%) compared with placebo. Twelve-month OS rates were 26.6% versus 10.9%. Subsequently, nivolumab has obtained a license in advanced gastric cancer in Japan, while licensing applications in the US and EU are ongoing. Separately, the first study targeting PD-1 in squamous cell carcinoma oesophagus with nivolumab was conducted and was unselected for tumour PD-L1 positivity. 18 An ORR of 17% and mOS of 10.8 months were observed. Of note, for the patients who developed investigator-evaluated immune-related toxicity, ORR was 25%.
Preclinical data have shown that the combination of PD-1 and CTLA-4 receptor blockade might improve antitumour activity. 19 This enhanced efficacy may be hypothesised to be due to alternative pronged approaches in targeting the cancer immunity cycle. 20 Indeed, even though the single agent CTLA-4 inhibitor studies had failed to show an improvement in survival compared with placebo, the authors of the ipilimumab phase II study argue that its tolerability supports its development in combination with other ICPIs. 15 In the CHECKMATE 032 study, both nivolumab monotherapy and the combination of nivolumab plus ipilimumab were tested in heavily pretreated patients with advanced gastric cancer. 21 CHECKMATE 032 allocated patients to nivolumab (3 mg/kg) monotherapy and two dose schedules of nivolumab plus ipilimumab, nivolumab 1 mg/kg and ipilimumab 3 mg/kg, or nivolumab 3 mg/kg plus ipilimumab 1 mg/kg. The ORR was 14% (nivolumab alone), 26% (nivolumab 1, ipilimumab 3), and 10% (nivolumab 3, ipilimumab 1). Six-month PFS rates were 18%, 24%, and 9%, and 12-month OS rates were 39%, 35%, and 24%, respectively. There was some correlation between ORR and PD-L1 expression (divided into <1%, 1–5%, and ⩾5%) but no linear relationship. In the nivolumab 1, ipilimumab 3 subgroup, the PD-L1-positive population seemed to derive more of an OS benefit: 12-month OS rate 50% (versus 35%). However, due to the increased toxicity observed with higher ipilimumab dosing in this trial 21 and others, many investigators would favour a lower ipilimumab dose. A first-line trial combination of ICPI (nivolumab + one of four doses of ipilimumab) followed by maintenance nivolumab versus the investigator’s choice of capecitabine/oxaliplatin (XELOX) or fluorouracil/leucovorin/oxaliplatin (FOLFOX) is recruiting [ClinicalTrials.gov identifier: NCT02872116].
After the phase I dose escalation trial of avelumab, an anti-PD-L1 antibody in advanced solid tumours, showed early evidence of efficacy, 22 with some isolated reports of meaningful clinical benefit in gastric cancer, 23 this drug has been further evaluated in the phase Ib expanded cohort JAVELIN study in two different settings. During second-line treatment, a similar ORR to that observed for nivolumab and pembrolizumab (15%) was observed. A first-line maintenance registration study is ongoing (JAVELIN Gastric 100) [ClinicalTrials.gov identifier: NCT026-25610]. Recently, however, a phase III trial comparing avelumab with standard of care chemotherapy has been reported by the trial sponsors not to show a survival improvement. 24 Of note, recent preliminary data of Keynote-061 in which pembrolizumab was compared to paclitaxel in the second-line setting, have also failed to demonstrate an improvement in either PFS or OS. 25 Full results of these studies are yet to be published, but in the context of the licensing of nivolumab and pembrolizumab in later lines of treatment, these data have nonetheless been provocative. It is not clear whether biological differences between anti-PD-1 and anti-PD-L1 inhibition translate into meaningful differences in efficacy between these classes of drugs. In a solid tumour phase I trial of another anti-PD-L1 antibody, durvalumab, activity was observed in gastric cancer, and several clinical trials are further investigating this compound in this disease. 26 PLATFORM is one such trial in which, if at least stable disease is demonstrated after first-line chemotherapy [ClinicalTrials.gov identifier: NCT02678182], patients receive one of four maintenance treatments, one of which is durvalumab. This randomised phase II trial has an adaptive study design with a plan to expand the effective arms into a phase III maintenance trial powered for OS. As more robust data become available for other biomarker-selected populations (e.g. hepatocyte growth factor receptor (MET) positive or Fibroblast Growth Factor Receptor (FGFR) amplified), it may be possible to amend the overall trial design to incorporate these biomarker-targeted maintenance therapies. Table 1 provides a summary of the pivotal phase III trials of ICPIs in advanced gastric cancer.
Pivotal phase III ICPI studies in OG cancers.
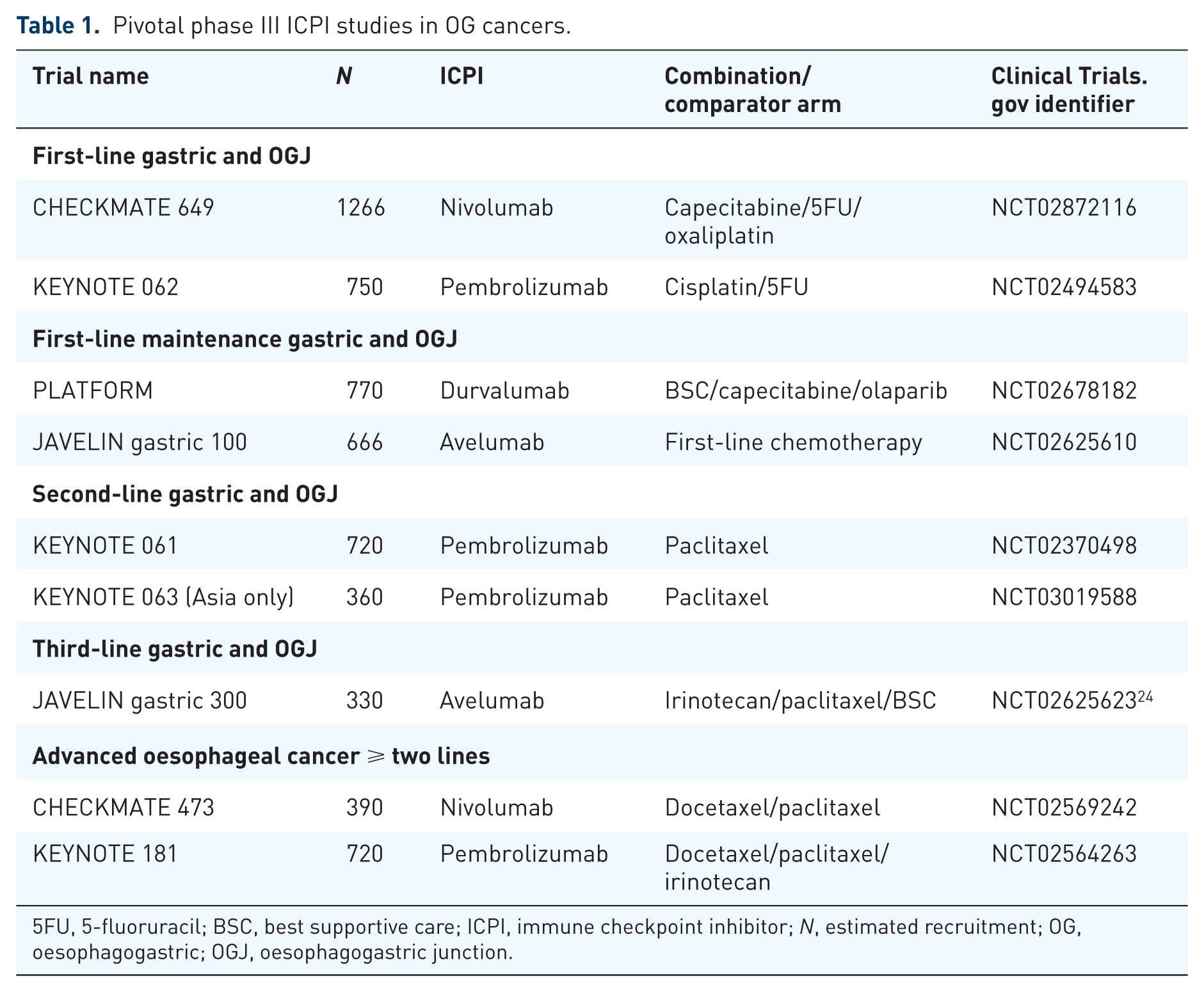
5FU, 5-fluoruracil; BSC, best supportive care; ICPI, immune checkpoint inhibitor; N, estimated recruitment; OG, oesophagogastric; OGJ, oesophagogastric junction.
Clearly, specific challenges remain in identifying biomarkers that may permit better prediction of those patients likely to derive benefit from ICPIs and, not least due to their high costs, the duration of treatment required to maintain responsiveness. In this review, we will explore the known and hypothesised mechanisms of resistance to and predictive biomarkers of response to ICPIs, and define strategies which may be used to enhance the efficacy and duration of response to these agents in advanced OG cancer.
Biomarkers of responsiveness to ICPIs and other immuno-oncology (IO) drugs in OG cancers
One approach to identifying biomarkers is the further molecular refinement of subtypes of gastric and oesophageal cancer. The search for new cancer genes has exploded through large-scale molecular characterisation initiatives such as The Cancer Genome Atlas (TCGA) and the Asian Cancer Research Group (ACRG). These types of characterisations may more ultimately explain why some tumours respond particularly well to ICPIs. Interestingly, in gastric cancer, although there are similarities between the TCGA and ACRG classification systems, only the ACRG seems to serve as a useful prognosticator. This prognostic difference between the TCGA and ACRG may be related to the limited follow up of the TCGA, the incorporation of clinical characteristics into the ACRG system and the inclusion of molecular aberrations in the ACRG system which are not mutually exclusive if applied within the TCGA system. 27 The TCGA also recently reported the genomic characterisation of oesophageal adeno and squamous carcinomas. 28 Other molecular categorisations of relevance include one which attempts to link the alterations to markers of potential therapeutic significance. 29 The categories which are defined by this system include enrichment for prevalent defects in the homologous recombination repair pathway, mutational patterns associated with a high mutational load and those associated with an ageing imprint.
In both oesophageal and gastric cancer, none of the defined molecular subtypes on their own have been shown to be predictive biomarkers for ICPIs. One reason for this may be the fact that most of these analyses have been conducted on early, resectable cancers, when in fact the situation in heavily pretreated advanced cancer is likely to be significantly different. Indeed, it seems likely that a multifaceted approach will be required in advanced disease. In the next sections, isolated biomarkers of relevance which may comprise a composite prognostic tool will be explored in some detail.
PD-L1 expression
Across oncology indications, many IO studies continue to use PD-L1 expression as a stratifying factor. However, the importance of PD-L1 expression in ICPIs in gastric cancer remains unclear. This is for composite reasons, including the variable sensitivity of the companion diagnostic used to quantify PD-L1 expression and the lack of specificity of the cell types on which it is being quantified between each of these companion diagnostics (Table 2). Most of these studies perform PD-L1 quantification by immunohistochemistry on archived tissue, providing only a snapshot of PD-L1 intensity and distribution in one part of the tumour, and may not be representative of the changing tumour microenvironment (TME) in other parts of the primary tumour or in subsequent metastases. 30 This PD-L1 data disparity clearly demonstrates that a more complex approach to biomarker identification is required.
Key examples of ICPI FDA companion diagnostic approvals.
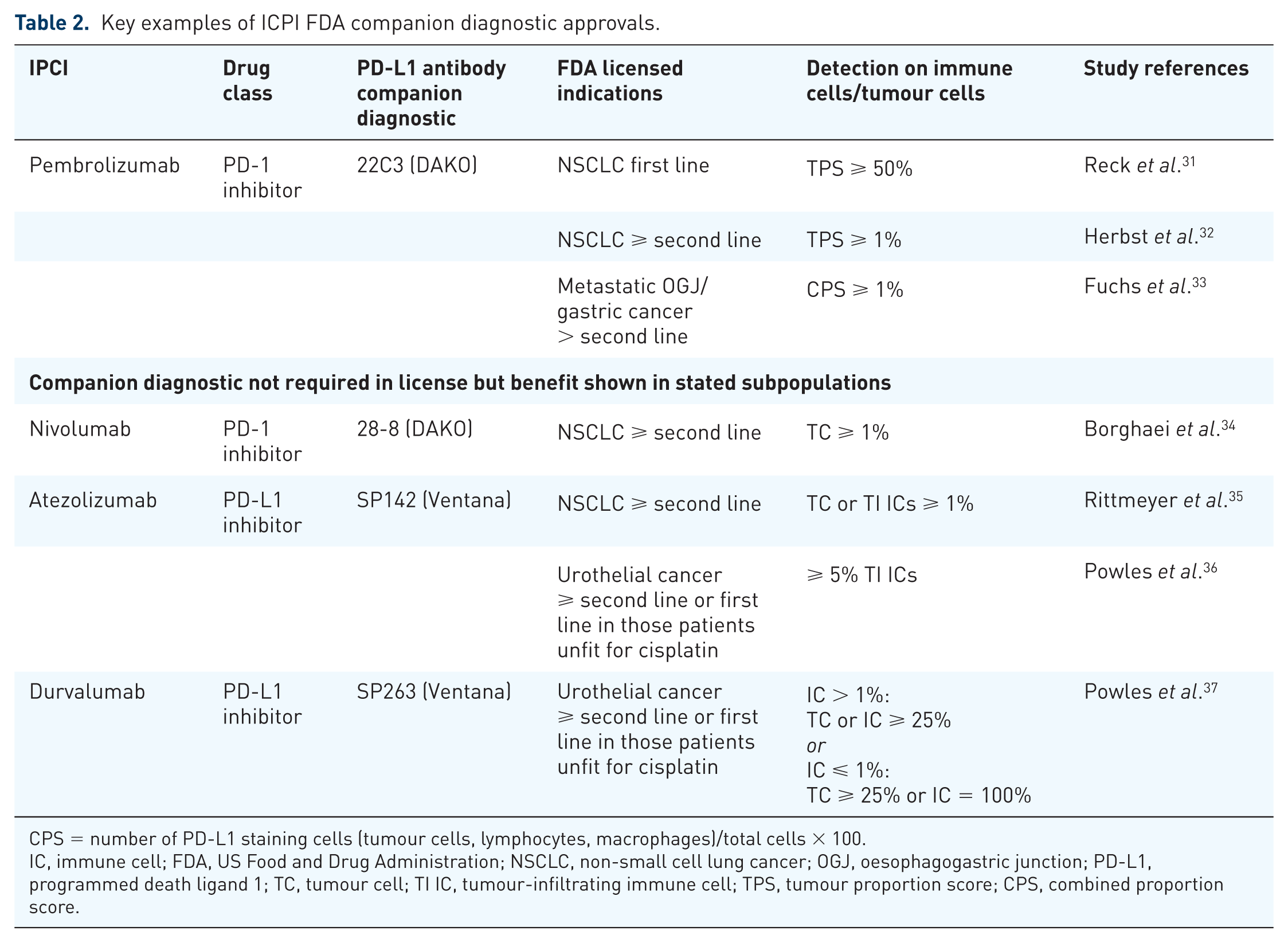
CPS = number of PD-L1 staining cells (tumour cells, lymphocytes, macrophages)/total cells × 100.
IC, immune cell; FDA, US Food and Drug Administration; NSCLC, non-small cell lung cancer; OGJ, oesophagogastric junction; PD-L1, programmed death ligand 1; TC, tumour cell; TI IC, tumour-infiltrating immune cell; TPS, tumour proportion score; CPS, combined proportion score.
Mismatch repair deficiency
Microsatellite instability (MSI) occurs as a result of defective mismatch repair (MMR). 38 Microsatellites are tiny, repetitive DNA sequences which accumulate mutations when MMR genes dysfunction or are silenced epigenetically. MMR deficiency leads to a hypermutated phenotype and high levels of neoantigen presentation, and is associated with an enhanced response to pembrolizumab across tumour types. 39 Consequently, the FDA granted pembrolizumab accelerated approval for MMR-deficient solid tumour indications, and was the first tumour agnostic approval of its kind in its history. In Keynote-012, two of the four patients with MMR-deficient gastric cancers demonstrated a radiological response. 16 Indeed, in detailed genomic analyses of MMR deficiency and its association with other immune factors, both MMR deficiency and tumour-infiltrating lymphocyte (TIL) presence were associated with better prognosis. Keynote-059 cohort 3 data in which patients with PD-L1 positive tumours (combined proportion score of ⩾1%) were enrolled to receive pembrolizumab in the first line was recently published. 33 Similarly, in this study, ORR values were dramatically better in patients whose tumours demonstrated high MSI compared with those patients whose tumours did not (57.1% versus 9%). It could be hypothesised that patients with these features may be more likely to respond to PD-1 targeting. 40
Tumour mutational burden
The utility of TMB as a prognostic biomarker is becoming increasingly evident across tumour types. The first detailed study examining whether a correlation between mutational burden and response to CTLA-4 blockade could be demonstrated was conducted in patients with melanoma. 41 Indeed those patients with a higher TMB had better responses to CTLA-4 inhibition and this provided a rationale for examining exomes of patients for whom anti-CTLA-4 agents was being considered. Similarly, higher TMB has been shown to predict a favourable outcome to PD-1/PD-L1 blockade across diverse tumours. 42 In addition, in the recently published anti-PD-1/CTLA-4 combination first-line study in NSCLC, PFS among patients with a high TMB was significantly longer with nivolumab plus ipilimumab than with chemotherapy. 43
Mechanistically, a high density of neoepitopes stemming from either somatic driver or passenger mutations may permit more ready immune detection after ICPIs, leading to destruction of cells bearing these mutation-associated neoantigens. 44 Routine analysis of neoantigens was not realistic until the recent evolution in high-throughput next-generation sequencing technology. The feasibility of using a targeted sequence panel as a surrogate for the whole-exome TMB has been tested 6 and is likely to become increasingly utilised. However, problems in the definition of thresholds of higher TMBs and whether TMB is expressed as total number of mutations/exome, or mutations per megabase differ between studies and make interstudy interpretation of its utility as a predictive biomarker difficult to ascertain. Apart from the definition of the number of mutations, subtypes of mutations may be relevant, including whether they represent frameshift insertion/deletion versus nonsynonymous single nucleotide variants. 45 Tumour mutational analyses are nevertheless becoming significantly refined 46 and are likely to permit more tailored treatment selection in future years.
Immune cell gene expression signatures
It has previously been shown that gastric cancer in Asian and non-Asian patients might exhibit distinct gene signatures related to inflammation and immunity. 47 In particular, immune T-cell expression signatures were enriched in non-Asian gastric cancers, including both CD28 and CTLA-4 signalling, with supportive immunohistochemistry data showing T-cell markers (CD3, CD45R0, and CD8) significantly enriched in gastric cancer in white patients compared with Asian patients. The exception was the immunosuppressive T-regulatory cell marker FOXP3, which was significantly enriched in the Asian population. Additionally, notable increased expression of the macrophage marker CD68 occurred in non-Asian patients. In multivariate analysis, only CD68 and CD3 expressions were independently associated with survival, and a high CD68/CD3 ratio was predictive of worse OS. These have not yet been investigated as predictive biomarkers. These immune-related differences were distinct and unrelated to Epstein-Barr virus (EBV) infection and MMR status. Interferon (IFN)-γ gene set enrichment was however more frequently seen in EBV-infected and MSI subgroups, although there was no association between IFN-γ signature and total number of mutations. 48 Although T-cell expression signatures are becoming easier to measure,49,50 their reliability as predictive biomarkers needs to be verified. Indeed, in the Keynote-012 study, the identified IFN-γ gene expression signature did not significantly predict response to pembrolizumab, 16 although larger-scale analyses may yet prove its value as a predictive tool. For example, the 18-gene T-cell-inflamed gene expression profiling score utilized in the Keynote-059 study did indeed demonstrate a higher score in aggregate for responders than for nonresponders. 33
Other potential biomarkers of interest
Helicobacter pylori
Apart from its well established aetiological role in gastric cancer, Helicobacter pylori inflammation occurring secondary to H. pylori infection alters the gastric microenvironment in multiple ways. H. pylori infection increases the levels of DNA methylation of tumour suppressor genes, and the induction of inflammation-related genes, 51 which may be responsible for inducing a T-cell response in gastric mucosa and increasing expression of PD-L1. It might therefore be hypothesised that H. pylori-driven tumours might be more likely to respond to ICPIs, although there is no randomised clinical evidence for this.
Epstein-Barr virus
Amplification of the 9p24.1 locus, which leads to PD-L1 overexpression, is observed in approximately 11–15% of EBV-positive gastric cancer cases.52,53 However, other mechanisms responsible for PD-L1 upregulation aside from PD-L1 gene amplification in EBV-positive gastric cancer might include IFN-γ induction of PD-L1 expression via activation of JAK2/STAT1/IRF-1 signalling in cancer cells. 54 EBV-infected cancer such as Hodgkin’s lymphoma may respond well to ICPIs. 55 However, limited clinical data are available on the interaction between EBV status and ICPIs in gastric cancer.
T-cell receptor repertoire and gut microbiome
The role of T-cell receptor clonality and diversity in predicting response to ICPIs is the subject of investigations in many laboratories. 56 The associations between microbiome diversity and nonresponse to ICPIs have also been recently described.57,58 It is becoming apparent that primary resistance to ICPIs can be due to specific gut microbiome profiles and that antibiotics which alter this profile may limit the clinical efficacy of ICPIs. In keeping with this, faecal transplantation from ICPI-responding patients into germ-free or antibiotic-treated mice improved ICPI antitumour efficacy. 58 The mechanisms by which specific enteric bacteria modify the immune response during immunotherapy remain largely obscure, although the cross reactivity between microbial and tumour antigens is thought to enhance immune cell priming. 59
Mechanisms of acquired resistance to ICPIs
Many patients who have had an initial objective response to ICPIs will go on to relapse despite continuous immunotherapy treatment. 60 Factors which predispose patients to acquired ICPI resistance may include the acquisition of defective mechanisms for tumour antigen presentation, which predominantly occurs via alterations in major histocompatibility complex (MHC) class I expression, genetic or epigenetic alterations within immunogenic signalling pathways, and a shift towards an immunosuppressed tumour microenvironment. 61 There is clearly a role for treatments which may target each of these acquired resistance mechanisms and enhance the likely benefit which could be derived from ICPIs.
The loss of β2-microglobulin (B2M) is an example of an established mechanism of acquired resistance to immunotherapy which results from defective antigen presentation.62,63 B2M loss is thought to specifically interfere with MHC class I heavy chain folding, leading to a loss of its receptor localisation and curtailment of downstream signalling. Another mechanism for resistance development may be altered oncogenic signalling of tumour cells, with the IFN signalling pathway being the most intensively studied. 64 During ICPI treatment, multiple inhibitory checkpoints might also be upregulated because of IFN signalling 65 and activation of various pathways 66 in tumour-infiltrating lymphocytes (TILs), ultimately leading to therapeutic failure. For example, T-cell immunoglobulin and mucin-domain containing 3 (TIM-3), an immune checkpoint, has been upregulated in lesions from patients with lung adenocarcinoma who initially had a partial response to anti-PD-1 therapy. 67 Alterations in the WNTβ-catenin signalling pathway, 68 signalling alterations resulting from either phosphatase and tensin homolog (PTEN) loss or phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PI3KCA) mutations in tumour cells 69 and other receptor tyrosine kinases including epidermal growth factor receptor, 70 may all contribute to an immunosuppressive phenotype. Resistance to ICPIs may also occur via alterations in the tumour immune milieu. Greater infiltration of immunosuppressive myeloid cells, including type 2 tumour-associated macrophages, has been shown to correlate with ICPI resistance. 71 Chemokines play an important role in optimal T-cell recruitment to tumours following activation of the stimulator of IFN genes (STING) pathway in dendritic cells (DCs)72,73 and curtailment in type 1 IFN activity will likely result in reduced inflammatory T-cell infiltration. Alterations in the activity of other chemokines, including CXCR4, may contribute to this immunosuppressive phenotype. Alternatively, metabolic processes within the TME have also been shown to reduce immunogenicity, including altered adenosine metabolism which pivots the immune milieu towards one favouring immune suppression. 74 Alterations in angiogenesis may also play a role in the development of ICPI resistance. 75 Figure 2 provides an overview of some of the changes that may occur in the TME during tumour progression, each of which contributes towards an immunosuppressive environment, T-cell anergy, and immune escape.

The shift towards an immunosuppressed tumour microenvironment (TME) during tumour progression. Tumour cells stimulate the recruitment of myeloid and lymphoid derived immune cells, including macrophages, CD4+ Tregs, and MDSCs, to the TME via secretion of key cytokines that are chemotactic for these cell types. Tumour-cell derived anti-inflammatory cytokines, TGF-β1, and macrophage colony-stimulating factor (M-CSF) induce macrophages, CD4+ T cells, and MDSCs to upregulate immune-suppressive molecules including IL-10, TGF-β1, and CTLA-4, as well as pro-angiogenic factors. This results in further immune suppression, altered metabolism, and angiogenesis in the TME, promoting tumour progression. CTLA-4, cytotoxic T-lymphocyte-associated antigen 4; IDO, indoleamine-2,3 dioxygenase; IL, interleukin; TGF, transforming growth factor; Treg, regulatory T cell; MDSC, myeloid-derived suppressor cells.
Role of combination of ICPI with chemotherapy, targeted therapies, radiotherapy and other immuno-oncology drugs
The success of ICPI treatment across solid tumours has initiated a flurry of research into optimal combination strategies. Some of the reasons for primary and acquired resistance against ICPIs have been outlined and perhaps provided a justification for how combination therapies may be used to circumvent these. The optimal treatment of OG cancers has very specific considerations of the most appropriate combination therapies which will be outlined in this section.
Immuno-oncology and chemotherapy or radiotherapy
The rationale for the enhancement of the antitumour effects of chemotherapies by their upregulation of immunogenicity is not novel. Some chemotherapeutics are thought to be particularly immunogenic, 76 inducing immunogenic cell death by the combination of exposure of danger-associated molecular patterns, their recognition by toll-like receptors, and subsequent DC activation. 74 Of relevance to gastric cancer, a 30-year-old trial which combined Vibrio cholerae neuraminidase (VCN) treated autologous tumour cells and bacillus calmette-guerin (BCG) with cyclophosphamide, mitomycin C and 5-fluorouracil, showed improved survival relative to historical controls, 77 suggesting a recognised potential benefit of harnessing the immune system to improve chemotherapy responsiveness in this disease. Of relevance in ICPI-resistant models, pretreatment with chemotherapy may in fact induce ICPI sensitivity. This is demonstrated in a mouse model of NSCLC, in which oxaliplatin plus cyclophosphamide pretreatment rendered anti-CTLA-4 and anti-PD-1 antibodies efficacious. 78 Although data in gastric cancer models are lacking, these NSCLC model data do support a role for oxaliplatin-enhanced immunogenicity. In addition, preclinical data in BRCA1-deficient triple-negative breast cancer models suggest that cisplatin may be utilised to increase mutational burden and enhance responsiveness to combination immunotherapy. 79 As a proportion of patients with gastric cancer harbour defects in homologous recombination repair, 80 an understanding of how platinum drugs may be utilised to enhance immunotherapy efficacy should continue to be sought.
Clinical efficacy data of combinations of immunotherapy with chemotherapy in OG cancer are evolving and perhaps the most relevant presented data in the advanced setting were demonstrated in the other cohorts of Keynote-059, in which pembrolizumab was given either with or without chemotherapy. 81 Cohort 1 patients received pembrolizumab alone after at least two prior lines of therapy. Cohort 2 patients received pembrolizumab and cisplatin and 5-fluorouracil or capecitabine in the first-line setting. Confirmed ORR was 12% in cohort 1 and 60% in cohort 2. Median PFS was two and seven in cohorts 1 and 2, respectively. Further large randomised trials comparing and combining immuno-oncology (IO) with chemotherapy are ongoing (Table 1).
Similarly to the immunogenicity that chemotherapy is thought to enhance, radiotherapy has been shown to be associated with upregulation of PD-L1 in the tumour microenvironment (Teng et al. 2014). 82 Indeed, the coadministration of ICPIs with radiotherapy seems to translate to an enhanced efficacy of radiotherapy given alone. There is thus at least one currently recruiting clinical study combining ICPI with radiotherapy in advanced OG cancer [ClinicalTrials.gov identifier: NCT02830594].
IO and targeted therapies
Targeted therapies have provided a basis for the revolution of personalised cancer medicine over the last 25 years. However, most patients will develop resistance to these agents as resistant clones develop strategies to overcome inhibition of the targeted driver mutation. In OG cancer, few targeted agents have shown superior evidence of efficacy over standard of care approaches.
Antiangiogenics
The VEGFR2 antibody ramucirumab is now an established therapeutic option in the second-line treatment of advanced gastric cancer, either as a single agent or in combination with paclitaxel.4,5 There is evidence that VEGFR2 pathway activation by VEGFA might suppress antitumour T-cell activation, including via blocking the maturation of and upregulation of PD-L1 on DCs and enhancing regulatory T-cell activity. 83 Preclinical data have demonstrated that low doses of the murine parent antibody to ramucirumab, DC101, reprogrammes the TME and potentiates the antitumour efficacy of ICPIs. 84 In a colon adenocarcinoma murine model, a synergistic inhibitory effect of DC101 with anti-PD-1 therapy was demonstrated. 85 Crucially, vascular normalisation with antiangiogenic targeted therapies has been shown to reverse immunotherapy resistance in patients. 86 To test the safety and early efficacy of the combination of an antiangiogenic agent with ICPI in gastric cancer specifically, a phase I study combining pembrolizumab and ramucirumab in PD-L1-unselected multitumour patient cohorts has been initiated [ClinicalTrials.gov identifier: NCT02443324]. The safety profile was favourable, allowing administration of each drug at full dose. 87 Some early efficacy data have demonstrated antitumor activity (7.5% ORR) in previously treated gastric adenocarcinoma.
Targeting the angiogenic pathway with vaccine therapy is also an approach associated with some activity in gastric cancer. A vaccine against HLA-A24-restricted human VEGFR1-1084 and VEGFR2-169 demonstrated that for those patients who had an immunological response to the VEGFR2-169 peptides, there was a statistically significantly improved OS. 88
An alternative target of angiogenesis is the matrix metalloproteinase family of proteins. Andecaliximab binds to MMP-9 and inhibits its enzymatic activity, resulting in the inhibition of extracellular matrix protein degradation. 89 Although there was preliminary evidence of efficacy in ulcerative colitis, 88 the phase III trial of this agent was terminated early due to perceived futility. However, what can be gleaned from the knowledge of the basal activity of this enzyme is the predicted inhibition of angiogenesis, tumour growth, and metastasis. Thus, a phase II clinical trial combining andecaliximab with nivolumab in oesophagogastric junction (OGJ/gastric adenocarcinoma has completed recruitment [ClinicalTrials.gov identifier: NCT02864381] and the results are pending. By a similar mechanism, the mesothelin inhibitor CRS-207 is being trialled in patients with advanced or metastatic OGJ/gastric adenocarcinoma in combination with pembrolizumab [ClinicalTrials.gov identifier: NCT03122548].
Poly ADP (adenosine diphosphate)-ribose polymerase (PARP) inhibition
A large-scale study of 10,250 cancer genomes across cancer types demonstrated that, in addition to breast, ovarian and pancreatic cancers, gastric cancer exhibits defects in DNA repair. 80 Specifically, 7–12% of gastric cancers in this analysis had defective double-strand DNA break repair. Other markers of correct to homologous recombination repair (HRR) deficiency may include loss-of-function ataxia-telangiectasia mutated (ATM) tumours, which are thought to occur in 16–22% of patients with gastric cancer. 90 Although a phase II combination trial of the PARP inhibitor (PARP-i) olaparib and paclitaxel had shown early signs of promising activity, especially in ATM-deficient tumours, 91 a randomised phase III trial of olaparib plus paclitaxel versus placebo plus paclitaxel followed by olaparib or placebo (GOLD) failed to show a significant survival benefit. 92 Ongoing genetic analyses of GOLD will likely yield reasons for this apparent discrepancy. In view of the fact that tumours with HRR deficiencies may be particularly sensitive to immunotherapeutics given the potentially high mutagenic burden in tumours with BRCAness, 93 it may be worth continuing to explore this avenue in gastric cancer. For example, BRCA1-deficient triple-negative breast cancer is known to be associated with a high TMB and high CTL infiltration. 79 The PARP-i talazoparib has been shown to increase the percentage of cytotoxic CD8+ T cells, B cells, and natural killer cells on a BRCA1-deficient ovarian tumour cell line grown in the peritoneal cavity of tumour-bearing mice. 94 These observations suggest that ICPIs in combination with PARP-i may have potential therapeutic benefit and an in vivo BRCA1-deficient ovarian cancer model established in an immune-competent host was used to show that an anti-CTLA-4 antibody synergised with the PARP-i veliparib, resulting in an OS benefit for these animals. 95 Consequently, at least one trial is investigating the PARP-i/immunotherapy combination in advanced solid tumours [ClinicalTrials.gov identifier: NCT02734004].
HER2 inhibition
The rationale for the combination of anti-HER2 antibodies with immunotherapies in HER-2-positive gastric cancer 96 is based on trastuzumab’s well established efficiency in inducing antitumour activity via mediating antibody-dependent cell-mediated cytotoxicity, 97 increasing tumour lymphoid infiltration and reducing T-regulatory cell activity. 98 Combining this ability to enhance adaptive immunity with ICPIs has been shown to demonstrate synergism in vivo. 99 The PANACEA trial [ClinicalTrials.gov identifier: NCT02129556] is evaluating the efficacy of pembrolizumab and trastuzumab in patients with trastuzumab-resistant, HER2-positive metastatic breast cancer (MBC). The trastuzumab emtansine antibody drug conjugate T-DM1 is also being evaluated in combination with atezolizumab in patients with HER2-positive MBC whose disease has progressed on trastuzumab and a taxane [ClinicalTrials.gov identifier: NCT02924883]. Another ongoing phase Ib/II clinical trial is investigating the clinical activity and safety of the combination of pembrolizumab with trastuzumab, capecitabine, and cisplatin [ClinicalTrials.gov identifier: NCT02901301].
IO/IO drug combinations
In the setting of metastatic melanoma and recurrence after prior anti-PD-1 therapy irrespective of prior response to PD-1 inhibition, an alternative ICPI approach, CTLA-4 inhibition, demonstrated efficacy in some patients. 100 This study highlights the potential benefit of alternative approaches in the unmasking of the immune system’s antitumour efficacy. In this section, we discuss the rationale for and evolution of newer strategies of combining immune system targeted therapies in oesophagogastric cancer.
Combinations of ICPI with other ICPI or T-cell agonists
There are a number of adaptive-designed, phase II studies either recruiting or being planned to combine different IO agents in all solid tumours, including gastric cancer. For example, FRACTION-GC (a phase II, Fast Real-time Assessment of Combination Therapies in Immuno-Oncology study in patients with advanced gastric cancer) is currently randomising patients between nivolumab plus ipilumumab versus nivolumab plus the anti-LAG3 (lymphocyte activation gene 3) antibody BMS-986016, with scope to add further IO compounds [ClinicalTrials.gov identifier: NCT02935634]. An alternative approach being tested in advanced solid tumours including gastric cancer combines the anti-PD-L1 antibody avelumab with a selection of T-cell agonists, utomilumab (4-1BB agonist monoclonal antibody (mAb)), PF-04518600 (OX40 agonist mAb), the combination of the latter two or PD 0360324 (macrophage colony-stimulating factor (M-CSF) mAb) [ClinicalTrials.gov identifier: NCT02554812].
Targeting the TME
Tumour development is associated with the generation of an immunosuppressive tumour milieu consisting of multiple cell types, extracellular matrix, and metabolic mediators. 101 Each of these components potentially represents a hurdle to CTLs and their antitumour immune responses. One example of a TME metabolic mediator showing promise is the potent indoleamine-2,3 dioxygenase (IDO) inhibitor epacadostat. The IDO family of heme-dioxygenases catalyses the conversion of tryptophan to kynurenine and other metabolites that drive maintenance of an immunosuppressive TME in many cancers. 102 IDO inhibition synergises with ICPIs in preclinical models in their activation of intratumoural CD8+ T cells. 68 Based on the provisional positive results of trials combining nivolumab with the IDO inhibitor epacadostat 103 and pembrolizumab, 104 a further phase I/II study is currently assessing the safety and preliminary efficacy of epacadostat in combination with durvalumab in subjects with selected advanced solid tumours, including gastric cancer [ClinicalTrials.gov identifier: NCT02318277]. Preclinically, A2AR inhibition synergises with ICPIs 105 and recruiting early phase trials in advanced solid tumours include the combination of durvalumab with the A2AR antagonist AZD4635 [ClinicalTrials.gov identifier: NCT02740985].
Adoptive transfer therapies
Amplification of patient derived T cells ex vivo followed by reinfusion has been successful in melanoma; although few studies have been conducted in patients with gastric cancer. 106 A 44-patient study assessed the efficacy of adoptive immunotherapy with patient-derived tumour associated lymphocyte lines either in conjunction with chemotherapy or with chemotherapy alone. 107 Median survival was 8.5 months for the control chemotherapy alone group and 11.4 months for patients treated with adoptive T-cell therapy plus chemotherapy, though this result was not statistically significant. Chimeric antigen receptors (CARs) are engineered antigen receptor proteins consisting of an antigen-binding region and T-cell receptor signalling domains. 108 With this technology, T cells are genetically modified to express CARs, expanded ex vivo and adoptively transferred to patients, acting to redirect T cells’ effector functions upon binding to antigens on tumour cells. They are quicker to produce, are not restricted by human leukocyte antigen (HLA) type, and react to a wider range of molecules, thus making them more practical than TIL transfer. Although most often mentioned in the context of haematological malignancies, CAR T-cell technologies are becoming more refined and are likely to play an increasingly significant role in the treatment of solid tumours. For example, a phase I study using CEA-targeted CAR T cells in carcino-embryonic antigen (CEA)-positive gastric cancer and other solid tumours is ongoing [ClinicalTrials.gov identifier: NCT02349724].
Vaccine therapies, gut microbiota and oncolytic viruses
T cells may also be honed to tumour cells by vaccination. The general purpose of a cancer vaccine is to expand or boost a patient’s tumour antigen-specific T cells. 108 Vaccine therapies seek to exploit cellular immune responses to cancer antigens. Such antigens may be delivered to the host immune system as peptides or via DCs, which when activated act as powerful immune activators, though this is rapidly ablated by CD8+ lymphocytes. Use of NY-ESO-1 peptide (which is expressed in gastrointestinal cancers including oesophageal cancer) vaccines led to T-cell responses and tumour regression in at least one study. 109 DCs pulsed with tumour cell antigens have produced some initial promising results in gastric cancer. In one early study, a gastrointestinal tumour overexpressed MAGE-A3 peptide pulsed DCs were able to induce specific T-cell responses and minor tumour regression in some patients. 110 However, to enhance the short-lived immune responses seen in these small case series, multiple peptide personalised cancer vaccines are being developed and tested in combination with adjunctive approaches. 111
A better understanding of gut microbiota diversity will permit the selection of approaches that will facilitate the development of adjunctive therapies, including appropriate antibiotic, probiotic formulations, or commensal antigens with molecular mimicry to tumour antigens. 61 In a similar vein to the immune responsiveness induced by peptide vaccination, oncolytic viruses are thought to enhance immunogenicity. Newcastle disease viruses (NDV)-D90 induce gastric cancer cell apoptosis by stimulation of p38 signalling, suppression of ERK1/2 and Akt signalling, and angiogenesis inhibition. 112 In vivo, orthotopic injection of NDV-D90 impaired tumour growth and induced intratumoural necrosis, and clinical trials of other oncolytic viruses including recombinant vesicular stomatitis virus are underway [ClinicalTrials.gov identifier: NCT02923466].
Conclusion and perspective
Survival increments in advanced OG cancers have plateaued in the last decade, underlining the unmet clinical need for innovations in treating this group of diseases. In the current era of demonstrated ICPI successes across solid tumour malignancies, trials of ICPIs in OG cancers have expanded and have thus far met with variable success. These cancers have traditionally been considered to be more challenging to treat with immunotherapy than some other malignancies because of their variable mutational burden and infiltrating proinflammatory T cells. However, the OG cancers of responding patients demonstrate particular immune and molecular characteristics that make them particularly susceptible to ICPIs, with new predictive biomarkers of responsiveness continuing to be discovered and refined. Indeed, development of an understanding of acquired resistance mechanisms to ICPIs by a shift towards translational approaches to genomic profiling will encourage recruitment of patients into biomarker-driven combination trials. In order to bypass primary resistance mechanisms and to curb the development of resistance to ICPIs, an explosion in the numbers of rational combination strategies has taken place. Dynamic collaboration between industry, academic scientists and clinicians will be needed to accelerate those combination strategies most likely to be transformative for our patients.
Footnotes
Funding
Professor David Cunningham and Dr Naureen Starling are supported by the Royal Marsden NIHR Biomedical Research Centre and Dr Kate Young is funded by the Royal Marsden NIHR Biomedical Research Centre.
Conflict of interest statement
Dr Naureen Starling has obtained honoraria from AstraZeneca and research funding from Bristol Myers Squibb. Professor David Cunningham has obtained research funding from AstraZeneca Bayer, Celgene, Merrimack/Medimmune, Merck Serono and Sanofi. Other authors declare no conflict of interest.