
Abstract
Metastatic castrate resistant prostate cancer (PCa) remains an incurable entity. In the era of immunotherapy, the complex PCa microenvironment poses a unique challenge to the successful application of this class of agents. However, in the last decade, a tremendous effort has been made to explore this field of therapeutics. In this review, the physiology of the cancer immunity cycle is highlighted in the context of the prostate tumor microenvironment, and the current evidence for use of various classes of immunotherapy agents including vaccines (dendritic cell based, viral vector based and DNA/mRNA based), immune checkpoint inhibitors, Chimeric antigen receptor T cell therapy, antibody-mediated radioimmunotherapy, antibody drug conjugates, and bispecific antibodies, is consolidated. Finally, the future directions for combinatorial approaches to combat PCa are discussed.
Keywords
Introduction
Worldwide, prostate cancer (PCa) is the second-commonest cancer in men. Despite a 51% decrease in PCa-related mortality from 1993 to 2016, 1 it remains the fifth deadliest cancer in males, and metastatic castration resistant PCa (mCRPC) is still incurable. In 2020, PCa is projected to cause an estimated 33,330 deaths in the United States (US), which highlights the pressing need for novel and effective therapies. Given the recent success of immunotherapy in numerous hematological and solid malignancies, this territory is being explored widely in the PCa arena.
However, the unique tumor microenvironment of PCa poses a significant challenge to the successful application of immunotherapeutic agents.
This review will discuss the current state of immunotherapy in PCa, as well as highlighting the strategies currently under investigation that can help guide the use of immunotherapy in PCa for the future.
Cancer immunity cycle
Chen et al. elucidated the key processes in generating an antitumor immune response in a stepwise fashion. 2 The neoantigens created by the tumor are first captured by antigen presenting cells (APCs) (e.g., macrophages or dendritic cells). These are then identified by cytotoxic T lymphocytes (CTLs) in alliance with major histocompatibility complex (MHC) class I and II as well as costimulatory molecules, resulting in priming and stimulation of T cells. This is followed by channeling and infiltration of effector T cells to the cancer site, ultimately leading to recognition and killing of the malignant cells. However, cancer cells have developed multiple escape mechanisms to break this cycle. 3 For example, tumors may lose the expression of surface antigens, or the costimulatory/MHC molecules that induce CTLs. Tumor cells may express immunosuppressive cytokines [e.g., transforming growth factor beta (TGF-β)], or molecules such as programmed cell death ligands (PD-L1/L2) that bind to programmed cell death protein 1 (PD-1) receptor on T cells and attenuate their response. The tumor environment may be enriched with regulator myeloid derived suppressor cells (MDSCs) and T cells that further subdue innate and T cell-mediated antitumor immune responses. Tumor-infiltrating lymphocytes (TILs) may also have an upregulated expression of CTLA-4, which competes with co-stimulatory molecule CD-28 to bind B7 on APCs, and negatively regulates T cell function. 2 Besides CTLs, natural killer (NK) cells are nature’s second defense mechanism against malignant cells, and cancer cells can even evade killing by NK cells by engaging inhibitory NK cell receptors via class I MHC molecules, thereby counteracting the activation signal. 4
Current immunotherapeutic agents attempt to counteract a variety of these mechanisms and affect different stages of the cancer immunity cycle. Vaccines help elicit the T lymphocyte response by upregulating the initial step of antigen presentation. PCa is a lucrative target for vaccines given the abundance of tissue-specific proteins that are exclusive to prostate, thus minimizing off-target side-effects. 5 Checkpoint blockade using anti CTLA-4 and anti PD-1 monoclonal antibodies is being rigorously tested in PCa. Chimeric antigen receptor (CAR) T cells are another way of overpowering immune suppressive mechanisms by supplying modified T cells that precisely target the cancer-associated antigen. 6 These immunotherapeutic approaches, as well as some others, will be discussed individually.
Vaccines
Use of vaccines to generate active immunity has long been explored in oncology. The major categories of vaccines that have been developed so far include: (a) dendritic cell-based vaccines (e.g., Sipuleucel-T and DCVAC); (b) vector-based vaccines (e.g., ProstAtak or PROSTVAC); and (c) the newer DNA/mRNA-based vaccines. Here, the evidence for and against the use of the common vaccines that have been evaluated in PCa thus far is discussed, as well as newer vaccination strategies that are currently being studied.
Dendritic cell-based vaccines
Sipuleucel-T
is the only immune therapy currently approved by the US Food and Drug Administration (FDA) and is specifically for mCRPC patients with no visceral metastasis, who have no, or minimal, symptoms. In fact, it was the very first vaccine ever to be approved for use in any cancer. Early evidence came from studies in the 1990s on mice, which showed that dendritic cells could be removed from the host, activated ex vivo with tumor-related antigens, and replaced in vivo, resulting in protection from tumor inoculation. 7 This is achieved by collecting peripheral dendritic cells (DC) from the patient via leukapheresis and incubating them with granulocyte-macrophage colony-stimulating factor (GM-CSF) and prostatic acid phophatase (PAP) fusion protein called PA2024.8,9 Primed DC collected after 36–44 h are then introduced into the patient to produce PAP-specific CTLs (Figure 1). Two phase III trials (D9901 and D9902A) did not demonstrate any significant difference in disease progression in the cohort treated with Sipuleucel-T, but did report a 4.3-month increase in the overall survival (OS) after a 3-year follow-up. 10 This led to the revolutionary IMPACT trial, which included men with minimally symptomatic or asymptomatic mCRPC, and subsequently resulted in FDA approval of Sipuleucel-T. OS was prolonged by 4.1 months in the treatment cohort, although the time to disease progression was not significantly affected. In all of the three phase III trials, men in the placebo group with disease progression were allowed to cross over to receive Sipuleucel -T. A study later showed that these men had a significantly longer median OS of 20 months as opposed to 9.8 months in men who never crossed over. 11 This raises the possibility that the IMPACT trial underestimated the survival effect, given that over 50% of the patients in the IMPACT trial crossed over to receive immunotherapy. Further results from the PROCEED Trial (NCT01306890) that ended in January 2017, have not yet been analyzed. Treatment with Sipuleucel-T was well tolerated across all three trials. There were other trials to test the efficacy of Sipuleucel-T earlier in the disease course, although such a use is not yet FDA approved. Fong et al. conducted one such phase II trial for its use in neoadjuvant setting. Presurgical Sipuleucel-T administration in men who were scheduled for radical prostatectomies showed no downstaging of the tumor when compared with pre-prostatectomy biopsies. 12 Two other phase II trials using Sipuleucel-T compared concurrent versus sequential administration with androgen deprivation therapy (ADT) in men with mCRPC; both trials failed to find a difference in either treatment arm, with primary end points being CD-54 upregulation and peripheral PA2024-specific T cell proliferation response, respectively.13,14 Multiple studies have been carried out for its use in biochemically recurrent disease. A small phase II trial was conducted with 18 patients who had biochemical recurrence [defined as rising prostate specific antigen (PSA) level after definitive surgery or radiation]. Slowing of PSA doubling time was seen in 13 of the 18 patients treated with Sipuleucel-T, but no decrease in absolute PSA levels was noted. 15 Another phase II trial conducted by Beer et al. in men with biochemical recurrence after definitive local therapy found significant slowing of PSA doubling time, but did not find a difference in the primary end-point of time taken to develop biochemical failure (determined by PSA = />3 ng/ml). However, the findings of this trial were limited by methodological shortcomings, with no confirmatory PSA measured after biochemical failure, and an arbitrary primary end point of PSA level defining biochemical failure. 16 The STAND trial examined a novel primary endpoint of measuring systemic T-cell-specific immune response. Despite an increase in immune-mediated factors, no difference in time to next intervention or time to metastasis was seen. 17 A limitation of this trial was that it did not include a pure ADT comparison group; hence, the contribution of Sipuleucel-T alone on clinical outcomes could not be evaluated. Therefore, current evidence does not recommend Sipuleucel-T in biochemically recurrent disease. Likewise, there is no data supporting its use in men with castration resistant PCa (CRPC) whose only evidence of disseminated disease is an increased serum PSA level, or in those with more advanced disease (visceral metastases, using steroids and/or narcotics).
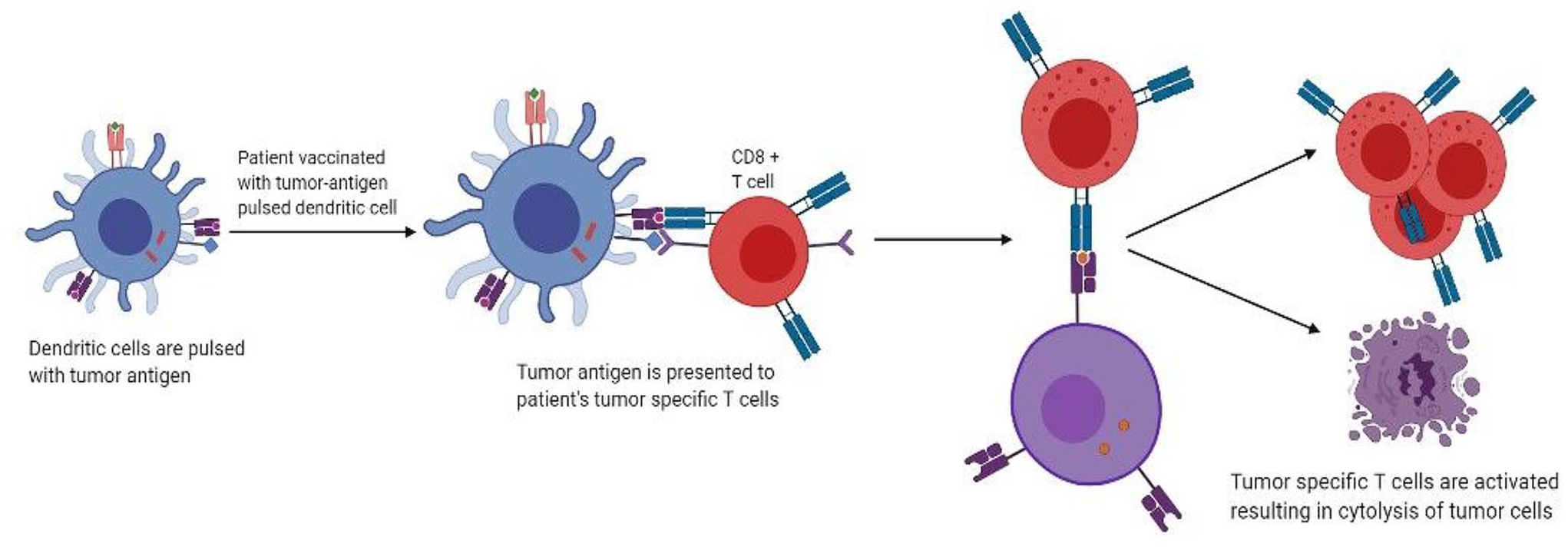
Mechanism of action of autologous dendritic cell vaccines.
A phase I/II trial showed that DCVAC/PCa resulted in 6–7 months OS advantage when given with prednisone and docetaxel in men with mCRPC (survival predictions values calculated by the Halabi and MSKCC nomogram). 18 However, another randomized phase II trial found the addition of DCVAC to docetaxel was not beneficial, with average follow up of 46.3 months despite the development of an immune reaction, which the vaccine was shown to cause, in these patients. 19 The VIABLE trial [ClinicalTrials.gov identifier: NCT02111577] is currently ongoing in mCRPC patients treated with prednisone and docetaxel versus prednisone, docetaxel, and DCVAC/P looking at OS, and is expected to be completed by June 2020. 20
Viral vector-based vaccines
An early phase I/II trial did not elicit any clinical benefit or reduction in PSA levels over a follow up of almost 4 years, despite demonstrating a substantially increased tumor infiltrating T cells compared with controls. 25 Absence of clinically meaningful outcomes instigated further investigation into treatments that combine standard therapies, such as hormonal therapy or radiotherapy with Adv-tk + GCV. The synergistic potential of ProstAtak and radiation therapy was evaluated in a phase I/II trial wherein patients with localized PCa (stage T1–T3) showed excellent locoregional control irrespective of high-risk features such as Gleason score >6 or pre-treatment PSA level >10 ng/ml, but those with pelvic lymphadenopathy did not attain systemic control. 26 Given the results of this trial, a phase III trial assessing ProstAtak in conjunction with radiotherapy for high and intermediate risk localized PCa patients is ongoing [ClinicalTrials.gov identifier: NCT01436968].
Activated CTLs then destroy PCa cells carrying the PSA antigen, subsequently allowing the immune system exposure to a range of novel cancer-associated antigens which then stimulates more CTLs for a deeper antitumor effect. This phenomenon is known as antigen spreading.28,29 Neutralizing antibodies can develop against a viral vector after the initial exposure to it, causing booster vaccination with the same virus (homologous prime/boost vaccination) to be unsuccessful. In order to bypass this problem, a unique strategy is the use of PROSTVAC–V/F regimen, wherein PROSTVAC-V (a recombinant vector derived from vaccinia) is administered first and then PROSTVAC–F is injected (a recombinant vector derived from fowl pox), in order to utilize a heterologous prime/boost strategy. This utilizes viral vectors derived from fowl pox and the booster vaccination, which both code for the same tumor-associated antigens. 30 This strategy was evaluated in patients with localized PCa with PSA recurrence after radical treatment. 31 A positive trend with prolongation of PSA-free survival was observed in the patient cohort that received a single injection of PROSTVAC-V followed by multiple injections of PROSTVAC-F. 31 Meanwhile, a study by Hodge et al. showed much greater T-cell activation when a triad of costimulatory molecules (TRICOM) – leukocyte function-associated antigen-3 (LFA-3), B7.1, and intercellular adhesion molecule-1(ICAM-1) – are used in conjunction with PSA. 32
GM-CSF in combination with PROSTVAC was investigated in a placebo-controlled phase II study in men with minimal symptoms and mCRPC with no history of chemotherapy. The trial demonstrated a 44% mortality reduction in patients who received this regimen. 33 However, the randomized double-blind phase III PROSPECT trial failed to show any impact on OS in mCRPC patients who received PROSTVAC-V/F, both with and without GM-CSF. 34 As a result, PROSTVAC as combination therapy in neoadjuvant/adjuvant setting with immune checkpoint inhibitors is under investigation [ClinicalTrials.gov identifier: NCT02933255]. 35 Similarly, concurrent use docetaxel and pox-virus-based vaccination has been proven to be safe and does not compromise the production of tumor-specific CTL-mediated immune response. 36 Results from a randomized phase II study also suggest that patients with non-metastatic, comparatively slow growing PCa may procure a survival benefit from the use of immunotherapy as front-line treatment or immunotherapy followed by antiandrogen therapy as opposed to antiandrogen therapy, or antiandrogen therapy upfront then immunotherapy as second-line treatment. 37 To further expand the scope of combination immune-therapeutics, PROSTVAC-V/F in conjunction with bi-functional fusion protein MSB0011359C and the CV301 vaccine (targeting PD-L1 and TGF-β) is being evaluated in men with recurrent disease after localized radical treatment [ClinicalTrials.gov identifier: NCT03315871].38,39
DNA/mRNA-based vaccines
Future of vaccines
Despite Sipuleucel-T being a harbinger of hope, subsequent vaccines in PCa have demonstrated only a lukewarm clinical response. New vaccine strategies utilizing novel viral and bacterial vectors, as well as combinatorial approaches, are being tested to expand the ambit of immunotherapy by vaccines. However, several challenges exist in optimizing response to vaccination. The immunosuppressive micro-environment of PCa is one such roadblock. Addition of immunometabolic agents such as Indoximod, which relieves suppression of mammalian target of rapamycin (mTOR) and thus restores normal effector T cell function or immune checkpoint inhibitors can potentially skew the balance towards an immunostimulatory tumor milieu. Radiotherapy has been proven to upregulate expression of TAAs and HLA as well as antigen processing molecules, and radiation-vaccination combination is being explored extensively in current clinical trials. 51 The role of androgen deprivation therapy in regulating immune response is well studied, is known to help in thymic regeneration, and can potentially render PCa cells immune-sensitive.52,53 Enzalutamide in combination with vaccine has shown in vivo benefit in mouse models with advanced PCa, and, if proven, to be effective in humans may represent a paradigm shift in management of CRPC. 54 Another question is the selection of appropriate target antigen to induce a formidable and sustained immune response. Genetic modification resulting in amino acid substitutions in native TAAs to generate “mimotopes” can trigger T cell cross-reactivity and generate a stronger immune response. 55 Targeting neoantigens produced by somatic mutations within the tumor can be another strategy, and has been trialed in malignancies with high mutational burden such as non-small cell lung cancer (NSCLC) and melanoma.56,57 Choosing an appropriate site and mode of delivery is an active research query as well. Biomaterials to synthesize stable, non-degradable, precisely sized and shaped particles that are readily taken up by APCs are being developed. There is some evidence that intra-lymph node injection may induce a better immune response compared with subcutaneous delivery, and this requires further exploration in clinical trials.58,59 Yet another approach can be using dendritic cell vaccines carrying mRNA derived from “cancer stem cells,” which has shown clinical response in glioblastoma. 60 While attempts are being made to develop more effective vaccines, several trials to unleash the full potential of vaccines in various combinations with other therapies are being conducted. Table 1 summarizes some of the ongoing trials.
Selected ongoing clinical trials in the treatment of PCa.

Ad5, adenovirus type 5; GM-CSF, granulocyte-macrophage colony-stimulating factor; mCRPC, metastatic castration resistant PCa; PCa, prostate cancer; PSA, prostate specific antigen.
Checkpoint inhibitors
Immune checkpoint inhibition (ICI) has revolutionized the field of cancer immunotherapy. Cancer cells can impair immune function, and specifically T-cell function, by expressing immune checkpoint molecules identified as self by the immune system, notably PD-1 and cytotoxic T lymphocyte antigen-4 (CTLA-4) with ligands PD-L1 and B7-1/B7-2, respectively. Therefore, ICI focuses on blocking these immune checkpoint molecules to enhance intrinsic anticancer immunity by the body (Figure 2). To date, anti-CTLA-4 therapy has shown reproducible responses only in malignant melanoma 61 In contrast, anti-PD-1 trials have been effective in a broad range of cancers, including NSCLC and Hodgkin’s lymphoma.62–64 To date, in urology, anti-PD-1 monoclonal antibodies (mAbs) have displayed a more favorable efficacy and safety profile as compared with second-line cytotoxic chemotherapy in renal cell cancer (RCC) and urothelial cell carcinoma. In platinum-pretreated urothelial cancer patients, pembrolizumab showed a 10.3 months OS versus only 7.4 months. Similarly, patients with advanced RCC who received nivolumab demonstrated a longer median OS (25.0 versus 19.6 months) and overall tumor response (25% versus 5%) as opposed to second-line Everolimus. 65 In this section, we discuss the roles, responses, and future therapeutic perspectives of CTLA-4 and PD-1/PD-L1 directed mAbs (ipilimumab and pembrolizumab/nivolumab, respectively).

ICI’s mode of action.
Cytotoxic T-lymphocyte associated antigen-4
CTLA-4 is a co-inhibitory signaling receptor expressed on CD8+ and CD4+ T cells. Instead of binding to the CD28 receptor (the primary receptor needed for activation of T cells), it binds to costimulatory ligands B7-1 (CD80) and B7-2 (CD86) on APCs, thereby impairing activation of T-cells. 66 Its constitutive activity on regulatory T (Treg) is known to contribute to their immunosuppressive capability, but its mechanism can be harnessed to produce antitumor responses with mAbs such as ipilimumab that specifically inhibit CTLA-4 and thereby dampen the immunosuppressive action of Treg cells. A variety of doses, schedules, and combinations of ipilimumab have been examined in PCa. Early trials with ipilimumab in PCa have confirmed that it has the same toxicity and safety profiles observed in other cancers, with just one dose of the drug (3 mg/kg) being safe and well-tolerated while also causing >50% reduction in PSA in 15–17% of mCRPC patients.67,68
Further, a phase III trial (CA184-043) for patients who had developed disease progression and bone metastasis despite docetaxel chemotherapy enrolled 799 patients who were treated with radiotherapy to bone followed by either ipilimumab or placebo. Although there was no difference between the groups for OS, the group that received ipilimumab demonstrated a longer PFS of 4 months versus 3.1 months on placebo [hazard ratio (HR) 0.70, p < 0.001]. Further subgroup analysis also indicated that ipilimumab might be more effective in patients with favorable disease features such as alkaline phosphatase <1.5× the upper limit of normal range, and hemoglobin concentrations of at least 11.0g/dl and no visceral metastasis. 69 For this subgroup of patients, median OS was 22.7 months [95% confidence interval (CI) 17.8–28.3] with ipilimumab versus 15.8 months (13.7–19.4) with placebo (HR 0.62, p = 0.0038). However, this survival benefit could not be extended to patients who had at least one high risk factor. The median OS for this subgroup was barely 6.5 months (5.7–7.9) with ipilimumab and 7.3 months (6.7–7.8) with placebo.
Combination therapy, in particular radiotherapy and ipilimumab, may also result in synergistic antitumor activity as radiation-mediated cell death can instigate the release of more tumor antigens that strengthen the immune-potential of ipilimumab. 70 A phase III (CA184-107) trial of ipilimumab given alone at 10 mg/kg in four doses 3-weekly or with external beam radiotherapy in mCRPC induced a PSA decline in 15% of patients. Further, other combinations such as ipilimumab with androgen deprivation therapy, GM-CSF, or vaccines have induced significant declines in PSA rates in 25–55% of patients with mCRPC.35,65,71 A higher CTLA-4/T-cell ratio has also been shown to confer worse prognosis, although combination phase I/II trials with ipilimumab and the cancer vaccine GVAX, have shown that high CTLA-4/T-cell ratios on peripheral blood smears may represent population subsets that are more likely to benefit from ipilimumab. 72
Although many trials did not reach their primary endpoints, the results remain promising, especially in the advanced castration-resistant patients, with the data suggesting that continued follow up of survival is warranted. While the efficacy of combination therapies is still not well known, ipilimumab should continue to be utilized until a better treatment regimen can be established.
PD-1/PD-L pathway
In contrast to CTLA-4, which inhibits an immune response during priming of T-cell activation, PD-1 acts as a negative checkpoint regulator during the effector phase, in both the peripheral tissues and the tumor microenvironment. In PCa, PD-1 is known to be expressed strongly on TILs, and is also associated with tumor progression.73,74 However, only some studies report a strong association with PD-1 expression and poor outcomes,73,75 with other studies reporting little association due to expression levels varying from 0% to 100%.76,77
A phase Ib study [ClinicalTrials.gov identifier: NCT02054806] with pembrolizumab (a humanized anti-PD-1 mAb) monotherapy for patients with PCa who had disease progression on standard therapy found that pembrolizumab elicited antitumor activity with a reasonable safety profile. Treatment effects lasted over 1 year in most patients (median = 13.5 months), with one patient even exhibiting stable disease for 20 months post treatment. 78 A recent study by Graff et al. also found a 30% response rate to anti PD-1 treatment for mCRPC, with evidence of progression on enzalutamide. 79 This suggests that the immune modulatory effect of androgen receptor inhibitors may be synergistic to immunotherapy. Yet, the recent phase III IMbassador250 trial [ClinicalTrials.gov identifier: NCT03016312] studying the combination of atezolizumab with enzalutamide versus enzalutamide alone in patients with mCRPC or incurable locally advanced PCa was terminated early as no difference in the OS, radiographic PFS or PSA response rate was observed between the two arms. 80 Median OS was 15.2 months for the combination arm versus 16.6 months for enzalutamide alone (HR = 1.12, p = 0.28). 80 One explanation for these results is that PD-L1 has low expression in PCa, and, therefore, may not be an ideal target. 81 Instead, PD-L2 was found to have a statistically significant correlation with immune-related pathways and prognosis, suggesting that PD-1 inhibitors such as pembrolizumab and nivolumab may also be acting through a PD-1/PD-L2 axis instead of PD-L1. 77 Targeting PD-L2 with external beam radiotherapy may be a possible combination for treating aggressive tumors in the future.
A combination ipilimumab and nivolumab phase II study [ClinicalTrials.gov identifier: NCT02601014] has demonstrated preliminary efficacy in asymptomatic or minimally symptomatic mCRPC patients, particularly in biomarker-enriched populations such as those with ARV7+ mutations. Results are especially promising in patients who had progressed after hormonal therapy but not yet received any chemotherapy. 82 Large-scale trials are required to clarify the impact of PD-1 blockade in PCa.
Adoptive cell therapy
CAR T cells are genetically engineered cells with receptors targeted against specific TAAs (Figure 3).

(A) CAR T cell interacting with a TAA presented in conjunction with an MHC molecule. (B) Schematic representation of a TCR and four generations of CARs.
CAR T cell therapy has taken the world of hematological cancers by storm, and its application is now being explored in solid tumors. PCa is a lucrative target for CAR-T cell therapy given the presence of multiple tumor specific antigens such as PAP, PSCA, PSMA, and PSA. 83
CAR-T cells comprise three main parts: an extracellular component that recognizes antigens, a transmembrane part as well as an intracellular component that mediates the final signal transduction. 84 Development in genetic engineering has led to tremendous improvisations in the structure of CAR-T cells, with four generations of CAR-T cells, each new one being a functional upgrade to its predecessor. Second-generation CAR-T cells differ from the first generation in possessing a costimulatory molecule such as CD28 or B7, which significantly enhances their cytotoxic ability. 85 Ma et al. constructed second-generation anti-PSMA CAR-T cells by inserting CD28 as a costimulatory molecule, which showed greater anti-tumor response in mouse models as compared with first generation anti PSMA CAR T cells. 86 They were shown to produce higher quantities of pro-inflammatory cytokines such as IL2 and IFNγ, with near disappearance of tumor in 3 weeks.
To further augment T cell proliferation, activity, and survival, a second co-stimulatory molecule was added to the third generation of CAR-T cells. The fourth generation of CAR-T cells, called TRUCKS (or CAR T-cells redirected for universal cytokine killing), increases the potency of CAR-T by adding a proinflammatory factor such as IL-2 to utilize the role of cytokines in tumor killing. Multiple phase I/II trials have been launched to assess CAR-T cell use in PCa. As suggested by Zhang et al., the use of PSMA-specific TGFβ-receptor dominant-negative autologous CAR-T cells can help overcome the immunosuppressive effect of TGFβ and intensify the oncolytic effect of CTLs. 87 A phase I trial evaluating this strategy in mCRPC patients is currently ongoing. 88 Phase I/II studies are also underway to investigate the feasibility and effectiveness of PSMA [ClinicalTrials.gov identifier: NCT01140373] as well as PSCA specific CAR T cells for the management of advanced PCa [ClinicalTrials.gov identifier: NCT03873805, NCT02744287].89–91
Yet another trial is evaluating EpCAM specific CAR T cells in EpCAM expressing malignancies. [ClinicalTrials.gov identifier: NCT03013712]. 92 With the exception of the [ClinicalTrials.gov identifier: NCT02744287] trial, these studies are expected to be completed between June 2020 and September 2021.
As attractive as it may seem, adoptive T cell transfer is not devoid of side effects. Cytokine release syndrome (CRS) has emerged as a potentially life-threatening complication of this therapy, with a frequency of >50%, reported in most recent clinical trials of CD-19 CAR T cells in hematological cancers. 93 CRS can manifest as mild flu-like illness, or can lead to a fatal systemic inflammatory response syndrome, with shock, acute respiratory distress syndrome, pancytopenia, acute renal failure, and multi-organ dysfunction. Utilizing a tumor specific antigen in CAR-T cells for PCa may theoretically carry less risk of systemic CRS, but clinical data is needed to substantiate this theory. CAR T cell-related encephalopathy syndrome is another complication reported in trials, and may present with focal neurological deficits or altered mentation. 94 Moreover, non-cancerous tissues that carry the targeted antigen can face inadvertent T cell-mediated damage. To overcome this issue, Kloss et al. suggested utilizing a combination of antigen recognition or “tumor sensing” to avoid off-target effects of T cells. 95 T cells are made to express dual receptors, one that does not allow for complete activation upon attachment of the respective antigen, along with a chimeric costimulatory receptor that binds to a second target antigen. Using PSMA and PSCA as two PSAs, Kloss et al. showed that such CAR-T cells only kill tissues that carry both antigens, but do not damage the tissues carrying either antigen alone. To solve the problem of the immunogenic nature of mouse antigens in humans and the large molecular size of scFvs, Hassani et al. used camelid nanobody (VHH) to engineer CAR T cells against PSMA, with promising results demonstrated in an in vitro model. 96 Thus, with future efforts targeted at enhancing the antitumor safety and efficacy profile, CAR-T cell treatment may be a novel and promising addition to our arsenal against PCa.
Antibody-based therapy
Over 100 years ago, Paul Ehrlich suggested using antibodies for selective killing of malignant cells, which was realized with the introduction of hybridoma technology in 1976, enabling the production of mAbs. 97 The FDA has currently approved mAbs for the management of numerous tumors, including lymphoma, leukemia, breast, colon, gastric cancers, etc. The presence of abundant target antigens makes PCa an ideal candidate for antibody-mediated treatment. mAbs act through multiple mechanisms; they can be conjugated to a chemotherapy or radiotherapy agent that focuses their cytotoxic effects by binding to specific target antigens, inhibits receptors that are key in progression and the onset of metastasis, or kills via both antibody dependent cell-mediated and complemented-mediated cytotoxicity. 98 PSMA and PSCA have been the two targets used most widely for such therapy. PSMA is a transmembrane epithelial glycoprotein found in both benign and malignant prostatic tissue and is targeted by J591, MLN2704, and CYT- 356 mAbs, whereas PSCA is a cell surface antigen found predominantly in malignant prostatic tissue, and AGS-PSCA is a completely humanized mAb targeting it.
Immunoconjugate therapy using mAbs
Immunoconjugate therapy utilizes an immune substance, such as a mAb that is chemically linked to a cytotoxic substance such as a toxin, radioisotope, or drug.
Radioimmunotherapy
A combination of radiation and immunotherapy (RIT), RIT involves using a small amount of radionuclide combined with a mAb, resulting in the formation of a radiopharmaceutical. mAbs recognize and bind to specific cell surface antigens, thus enabling the delivery of high amounts of radiation specifically into the tumor and minimizing off-target effects. Though a promising idea, RIT has shown consistent results only in hematological and lymphoid malignancies. However, PCa is unique among solid tumors as it is radiosensitive, expresses highly specific antigens that are easily targetable, and even a small volume of metastatic disease in bone marrow and lymph nodes can be picked up easily on imaging and accessed by circulating antibodies. 99 These properties can help PCa surpass the usual hindrances to application of radioimmunotherapy in solid malignancies.
As early as 1996, Deb et al. conducted a small phase I trial in a group of hormone-refractory mCRPC patients using 90Y-CYT-356 mAb. 100 However, none of the participants responded based on PSA level or imaging assessment. A phase I trial using 177lutetium-labeled J591 (another IgG1 monoclonal antibody targeting the PSMA external domain) in androgen unresponsive PCa, showed somewhat more promising results, with 11% patients (4/35) achieving >50% decline in PSA lasting for 3–8 months and 46% (16/35) showing stable disease lasting between 1–21 months. 101 In a pilot trial studying unlabeled versus radionuclide labelled J591 mAbs, Morris et al. demonstrated that antibody-dependent cell-mediated cytotoxicity (ADCC) was directly proportional to the antibody mass or dose. They further outlined that a dose of 100 mg was optimal if the intent was for immunotherapy, but optimal antibody mass seems to be 25 mg when used for the purpose of targeted radiotherapy. 102
In a similar study, Yttrium-90-labeled J591[(90)Y-J591] was used in men with CRPC. Only 2 of the 29 patients in this study experienced objective measurable disease response, which was reflected by a >50% fall in PSA levels, which lasted for about 8 months, whereas 6 patients (21%) were reported to have stable PSA levels. 103 Both the previously mentioned trials showed successful targeting of skeletal and soft tissue metastatic disease in most patients.102,103
Further, this evidence was supported by a phase II study by Tagawa et al. of lutetium-labelled J591 mAbs in patients with mCRPC. 99 The study revealed a dose response relationship as the 70 mCi/m2, that is, the higher dose cohort had statistically significant improved OS as compared with the 65 mCi/m2 cohort (21.8 months versus 11.9 months). They also demonstrated a significant decline, that is, >50% in circulating tumor cells in the higher dose cohort. Similar to prior phase I studies, 10.6% patients achieved >50% decrease in PSA levels, whereas almost 60% men demonstrated some level of decrease in PSA levels after treatment. Any decline in PSA level was significantly associated with improved survival, thus validating the role of PSA in monitoring response to treatment. The major dose-limiting toxicity, seen almost universally in all the trials, was myelotoxicity.
Antibody-drug conjugate therapy
Another novel agent is MLN2704, an immunoconjugate that utilizes the mAb MLN591 targeted against PSMA for intracellular delivery of maytansinoid-1(antimicrotubule agent) into PSMA-expressing cells. Galsky et al. conducted a phase I trial of MLN2704 on progressive mCRPC patients, wherein two (22%) of the nine patients sustained >50% decline in PSA compared with baseline. 104
However, a recent phase I/II dose escalation trial by Milowsky et al. of 62 patients with progressive mCRPC failed to show disease regression in any of the patients, with only 5 (8%) achieving >50% decline in PSA. Unfortunately, 71% patients suffered from peripheral neuropathy, forcing 38% of participants to stop the therapy. This narrow therapeutic window and unfavorable safety profile might be explained by linker lability and rapid deconjugation.
MLN2704 has been engineered by attaching an antimicrotubule protein (DM1) to a larger MLN591 antibody using a thiopentanoate link that contains disulfide bonds; however, these disulfide linker bonds are particularly labile, resulting in deconjugation of the bond and release of DM1 in the plasma itself rather than the targeted cancer cell, leading to considerable toxicity. This can be overcome by using immunoconjugates compounded with a less labile thioether linker. 105
Nejadmoghaddam et al. evaluated PLAC-1 (placenta specific-1) as another potential target for antibody-drug conjugate therapy against PCa in mouse models. 106
The antibody has demonstrated high affinity against PLAC-1 with rapid internalization after engagement with targeted lysis of PLAC1-expressing PCa cells only and improved efficacy of the cytotoxic agent, thereby suggesting the role of PLAC1 as an emerging immunotherapeutic target.
Antibody-dependent cell-mediated cytotoxicity
The ADCC mechanism has been explored in the form of anti-PSCA mAbs: AGS-PSCA and AGS-1C4D4. The first ever in human trial by Morris et al. enrolled 29 men with mCRPC; however, none of the patients achieved >50% PSA decline or a radiographic response. 107 Possible explanations for this are inability of the antibody to achieve optimal serum concentration or absence of PSCA expression in these PCa cells. As was shown in preclinical models, the naked antibody may not induce ADCC, calling for the development of chemoconjugated or radioconjugated antibodies. Antonarakis et al. conducted another phase I rapid dose-escalation trial of AGS-1C4D4, also a mAb against PSCA, in 13 patients with CRPC. 108 This study also failed to show >50% PSA reduction or an objective tumor response, although 46% (6/13) patients achieved stable radiographic disease lasting ⩽3 months. Both AGS- PSCA and AGS-1C4D4 had satisfactory safety profiles with no grade 3/4 toxicity.
Bispecific antibodies
An exciting and upcoming focus in immunotherapy is the production of genetically engineered Bi-specific antibodies, which serve not only as an effective link between the attacker (e.g., CTLs or radionuclides) and the target (e.g., cancer cells) but also interrupt two distinct oncogenic mediators. 109 In a pilot phase I study, eight mCRPC patients received CTLs along with anti-CD3 x anti-Her2 bispecific antibody (Her2Bi) and low dose IL-2 plus GM-CSF. 110 One patient showed partial response, while three out of seven patients had a substantial PSA decline as well as a significantly improved subjective assessment of pain.
Objective evaluation showed a rise in the levels of interferon gamma (IFN-γ) and T helper cell type 1 (TH-1) cytokines in peripheral blood mononuclear cells of two participants post treatment. With these encouraging results in mind, a phase II trial of pembrolizumab and HER2Bi armed ATCs in mCRPC patients is ongoing. 111
Another class of bispecific Abs, called BiTE or bispecific T cell engagers, has been developed to target PSMA. Anti-PSMA × anti-CD3 BsAb recognize CD3+ T cells and tumor cells expressing PSMA, promoting the cytolytic action of T cells. The first-in-human phase I clinical study of anti-PSMA x anti CD-3 diabody BAY2010112 was completed recently, with results yet to be published; another trial evaluating MOR209/ES414 in mCRPC patients is currently underway.112,113
A unique challenge in the use of antibody-based therapies that target PSMA in a mouse mode is their limited serum half-life. 114 Antibody-based immunotherapy devised using synthetic DNA plasmids that encode a therapeutic human mAb can help overcome the problems related to short serum half-life of mAbs and the need for frequent administration. 114 Similarly, chemical coupling with polyethylene glycol, fusion with heavy chain fragments or albumin are being tested as potential strategies to overcome the short half-life of bispecific antibodies. 115
Complex tumor microenvironment of PCa
Given the dynamic nature of immune cell types, the impact of the immune system on PCa is remarkably complex. Because high-grade PCa is characterized by low-level tumor infiltration of lymphocytes,12,116 the interactions between innate and adaptive immunity are not well understood. 117 Macrophages and Treg cells have been associated with aggressive pathology, high rates of recurrence after prostatectomy, and worse distant metastatic-free survival.77,118,119 On the other hand, mast cells, NK cells, and DCs are negatively associated with tumor progression,120,121 and have been shown to confer improved distant metastatic-free survival. 77 TILs in PCa may be dysfunctional and not capable of producing an immune response as suggested by examination of over 1500 resected PCa specimens wherein a greater TIL population was associated with a lower metastasis-free survival. 77 Low tumor-associated antigen expression, DNA mismatch repair gene defects, subdued expression of MHC class I, lack of phosphatase and tensin homolog (PTEN) protein, and poor IFN1 signaling are some key processes that play a role in this complex tumor environment. 122 One study revealed that the average mutation frequency in PCa is almost 10 times lower than melanoma, which may explain the significant difference in response to immunotherapy between the two malignancies.
Recent preclinical trials have shown that treatment with IFN-γ as well as radiation therapy, enhance MHC class I protein expression and may be associated with improved survival.123–125 The PTEN gene, which is known to be deleted in up to 30% of PCa cases, may be another emerging marker to assess immune responsiveness of PCa. 126 For example, melanoma patients with PTEN loss have a much greater treatment response to PD-1 blockade. 127 On the other hand, mutations in DNA damage repair (DDR) genes may in fact accord a survival benefit to patients treated with poly(ADP-ribose) polymerase (PARP) inhibitors such as Olaparib. 128
Nevertheless, most studies so far have had heterogeneous cohorts and small sample sizes, so the relation of tumor-immune interaction and clinical prognosis remains difficult to characterize. Future trials evaluating the impact of pre-immune status, IFN-γ and TGF-β signaling, MHC class I expression, PTEN and DDR gene mutations are needed to better understand the clinical implications of these factors in response to immunotherapy.
Future perspectives
‘Cold’ or immunosuppressive complex tumor microenvironment (TME) of PCa has been cited as a major cause for lack of success of immunotherapy hitherto. Immunomodulation by tilting the balance towards pro versus anti-inflammatory cytokines is a strategy that needs further exploration to overcome this roadblock. For example, TGF-β deletion in DCs has been shown to generate an immune response via suppressed Treg induction, whereas IL-2 can lead to T-helper and macrophage activation. 129 Indoximod, an indole-amine 2,3-dioxygenase inhibitor, can also convert the TME into an immune-permissive state by suppressing the effect of MDSCs, and this approach is under investigation in a clinical trial along with Sipuleucel-T. 130
Though not treated as traditional immunotherapy, radiation and chemotherapy also release neoantigens by tumor cell killing that leads to cross priming of additional antigen-specific T cells. This suggests a possible synergistic role of combining traditional antineoplastic agents with immunotherapy. 131 TGF-β neutralization combined with PD L-1 inhibition, development of TGF-β insensitive CAR-T cells, combining anti-PD-1 and anti-CTL4 antibodies or concurrent use of vaccination with checkpoint blockade, etc., are all combined approaches to augment multiple steps in the tumor immunity cycle.6,87,131
The other major barrier in immunology is the lack of an understanding of the true predictors of response. The traditional biomarkers of high tumor mutational burden and PD-1 expression are not seen consistently in PCa, necessitating the discovery of other biomarkers that can be utilized for cancer prognosis. Microsatellite instability was detected on biopsy of up to 54.5% of patients responsive to anti-PD-1 therapy, pointing towards an association between microsatellite instability and treatment response.132,133 Currently, the FDA has approved pembrolizumab for all unresectable and metastatic solid tumors displaying high microsatellite instability and mismatch repair deficient tumors, independent of histology or tumor site. This is due to the shared histopathological characteristics such as somatic hypermutation, neoantigen burden, and lymphocytic infiltration in tumors with high microsatellite instability. 134
Further, studies have found that combined T-cell immunoglobulin and mucin-domain containing molecule-3 (TIM-3) and PD-1 targeting was more effective in controlling tumor growth than either pathway alone, suggesting the role of both biomarkers as potential combined targets for therapy. 135 The role of radiomics in this contexture also needs to be evaluated. Interestingly, a recent study found that 3’-deoxy-3’-F-fluorthymidine positron emission tomography after 12 weeks was more predictive of PFS than changes in tumor size detected in computed tomography scan or PSA changes after 12 weeks. 136
Conclusion
Since the approval of Sipuleucel-T in 2010, no other immunotherapeutic agent has been approved by the FDA for use in PCa. However, tremendous research efforts are currently targeting multiple immune pathways, exploring combination strategies, using immune-intensification and immune-modulation, as well as developing new biomarkers for better stratification of the target population. As a result, immunotherapy might emerge as a viable tool in combating PCa. Currently, combination trials of ICI, vaccines, hormonal therapy, chemotherapy, radiotherapy, and PARP inhibition, as well as trials evaluating CAR-T cells and antibody-based approaches are all underway. As evidence accrues, we will be in a better position to decisively assess the clinical impact and the fate of immunotherapy in PCa.