
Abstract
Background:
A novel formulation of oral testosterone undecanoate (TU) was studied in a long- and short-term phase III trial to evaluate safety and efficacy.
Methods:
Hypogonadal men (age 18–65 years; two morning serum testosterone (T) <300 ng/dl with signs/symptoms) were recruited into a 365 day (trial I) or 105 day (trial II), randomized, multicenter trial. Patients were randomized 1:1 to oral TU (n = 161) or T-gel (n = 160) in trial I, and 3:1 to oral TU, twice daily (BID) JATENZO® (n = 166) or a topical T product [Axiron® (n = 56)] in trial II. Dose adjustments were based on average T concentrations (Cavg). Efficacy was assessed based on T levels, body composition and bone density. Safety was assessed by standard clinical measures.
Results:
Oral TU efficacy (% of patients with eugonadal T Cavg) was 84% (serum Cavg = 628 ± 343 ng/dl) and 87% (serum T equivalent Cavg ≈ 489 ± 155 ng/dl) in trials I and II, respectively. Oral TU significantly (p <0.0001) improved all Psychosexual Daily Questionnaire parameters in trials I and II. In trial I, lean mass increased 3.2 ± 2.7 kg and fat decreased by 2.4 ± 3.6 kg (both p <0.0001) and bone density improved in hip (+0.012 ± 0.0225 g/cm2) and spine (+0.018 ± 0.0422 g/cm2) after 365 days (both p <0.0001). Oral TU-associated adverse effects were consistent with other T-replacement therapies but oral TU patients experienced a greater number of mild gastrointestinal adverse effects. Oral TU subjects in both studies exhibited an increase in mean systolic blood pressure of about 3–5 mmHg. Oral TU was not associated with liver toxicity nor did it cause an elevation in high-sensitivity C-reactive protein or lipoprotein-associated phospholipase A2 (cardiovascular safety biomarkers) after 365 days of therapy.
Conclusion:
A new oral TU formulation was safe and effective and represents a significant therapeutic advance for the treatment of appropriate hypogonadal men.
Introduction
Testosterone (T)-replacement therapy (TRT) has evolved over time to provide healthcare providers and their hypogonadal patients with numerous treatment options. Beginning with early use of implanted T-pellets to injectable T-esters to oral methyltestosterone to a first-generation oral T-undecanoate (TU) product to scrotal and non-scrotal T patches and then to topical T-gels, the number of TRT choices continues to evolve. 1 More recent additions to the TRT armamentarium include a buccal patch, a long-acting T-undecanoate injection (intramuscular) product, a short-acting T-enanthate injection (e.g. 7 days; subcutaneous) and a nasal T-gel. Each of these delivery routes are associated with well-known drawbacks, including pain of injection, dermal irritation, T transference and potentially serious liver toxicity (e.g. oral methyltestosterone). In addition, dose adjustment to individualize patient response to these TRT methods can be challenging. Missing from the healthcare professional’s stable of TRT products is an oral T formulation that meets the current rigorous regulatory standards for safety and efficacy.
Historically, efforts to administer oral T have taken two primary paths: alkylation of T at the C-17 position to create T analogs that are resistant to first-pass hepatic metabolism (exemplified by methyltestosterone); 2 or fatty-acid esterification of T to create a T-ester (exemplified by TU) that is absorbed via the intestinal lymphatic system thus bypassing the portal circulation. 3 Oral methyltestosterone, originally discovered and used clinically in the mid-1930s, 1 is the only oral TRT ever approved for use in the US, but has been associated with serious hepatotoxicity such as cholestasis, peliosis hepatis, and hepatic adenocarcinoma4–6 and therefore is not recommended for clinical management of male hypogonadism. Conversely, while oral TU has not been associated with liver toxicity, an early oral TU formulation approved for use in many countries but never in the US (Andriol®) was highly influenced by dietary fat, thus leading to significant intra- and inter-patient variability in T response and questionable clinical utility.7,8 Reformulation of this product to reduce the effect of dietary fat did not address the low TU content of the capsules, thus resulting in the need to dose hypogonadal men with several capsules three or more times daily. Even then, reported serum T response would not result in average serum T levels in the normal range 9 and therefore would not pass current-day regulatory scrutiny for efficacy. Consequently, these oral TU formulations have never been widely used to treat T deficiency although they remain available in many countries.
To address the absence of an oral TRT product that meets current-day regulatory requirements for efficacy and safety, TU was formulated in a unique self-emulsifying drug delivery system (SEDDS) that was initially evaluated in short-term clinical studies. 9 SEDDS formulations combine hydrophilic and lipophilic excipients that enable the solubilization of highly lipophilic molecules like TU in the gut so that they may be absorbed after oral ingestion with a typical meal (no high-fat content required). 10 As depicted in Figure 1, absorption of oral TU occurs almost exclusively (>97%) via the intestinal lymphatic system, thereby bypassing the liver.3,11 Once in the circulation, endogenous non-specific esterases cleave T from the parent TU pro-drug. Each undecanoic (U) acid molecule (a straight-chain C-11 fatty acid) is metabolized by beta-oxidation to yield several molecules of acetyl-coenzyme-A and a single molecule of propionyl-coenzyme A. An unexpected finding during development of this new oral TU formulation was the degree of enzymatic cleavage of T from TU that can occur during blood-sample handling. This can lead to T assay values that do not reflect the actual circulating T concentrations; that is, they are artefactually high.12,13 Therefore, to determine efficacy based on the most accurate T concentrations in blood after oral TU, post-collection conversion of TU to T was minimized in men dosed with oral TU in the pivotal clinical trial described herein by assaying for T in plasma derived from blood collected into NaF-EDTA (sodium fluoride-ethylenediaminetetraacetic acid) tubes that were held on ice prior to centrifugation (a process that halts essentially all TU to T conversion). Additional studies (not detailed herein) were conducted to derive a correction that enabled the conversion of the average T concentration (Cavg) values attained from NaF-EDTA plasma into approximately equivalent serum T concentrations [In essence (due largely to a matrix effect), LC/MS-MS assay of T in NaF-EDTA plasma from men treated with oral TU yields a T value that is roughly 20% lower than a matched serum T value].
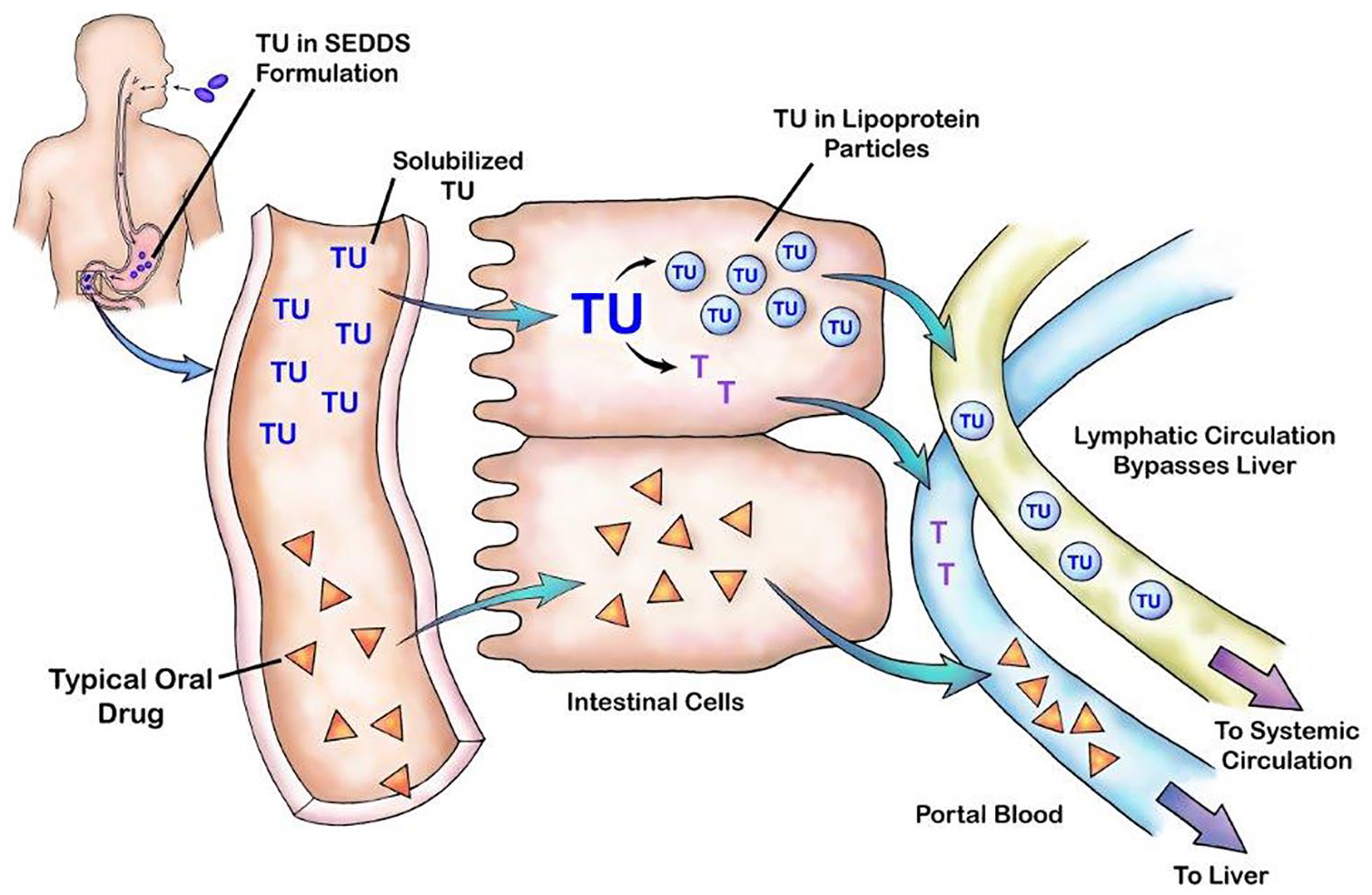
Pictorial representation of TU lymphatic absorption after oral delivery in SEDDS formulation.
This was important for two reasons. First, real-world clinical monitoring of T is based on serum T measurements. Second, it was necessary to adapt the dose-titration algorithm utilized in the pivotal trial of JATENZO (where T was assayed in NaF-EDTA plasma) for use with a single serum sample derived from blood collected into a standard plain collection tube (i.e. without added chemicals).
The present article summarizes two phase III clinical trials conducted to demonstrate long- and short-term safety and efficacy of a new oral TU formulation (JATENZO®) studies CLAR-09007, trial I [ClinicalTrials.gov identifier: NCT01403116] and CLAR-15012, trial II [ClinicalTrials.gov identifier: NCT00272278]. The primary difference between these studies was the starting oral TU dose and subsequent dose-titration algorithm employed to maximize the percentage of patients who achieved T Cavg within the eugonadal range without unacceptable peak T concentrations (Cmax). Additionally, in trial II, we evaluated T responses based on T concentrations in NaF-EDTA plasma, while serum T measurements were used in trial I that, in retrospect, yielded T concentrations that were artefactually elevated (by approximately 20%) due to post-collection of TU to T during blood-sample processing.12,13
Materials and methods
The phase III clinical trials detailed herein were approved by a central or site-specific institutional review boards before study initiation at each clinical site and were conducted in accordance with the Declaration of Helsinki and/or all relevant federal regulations, including good clinical practice guidelines. Written informed consent was obtained from trial participants before any study-related procedures were conducted.
Patient populations
Eligible patients were men aged 18–65 years, body mass index <38 kg/m2, with hypogonadism as defined by verified low morning serum total T <300 ng/dl (blood samples collected between 0600 h and 1000 h on 2 separate days approximately 7 days apart) and a history of signs and/or symptoms consistent with hypogonadism. Patients were naïve to androgen-replacement therapy or had an adequate washout of previous androgen-replacement therapies. Patients were excluded if they had significant uncontrolled intercurrent disease of any type, hematocrit (Hct) <35% or >48%, history of polycythemia, untreated, severe obstructive sleep apnea, abnormal digital rectal exam, prostate-specific antigen (PSA) >4.0 ng/ml, International Prostate Symptom Score >19 or history of prostate cancer. Prohibited medications included those that could affect T levels, T metabolism, or levels of T metabolites (e.g. antiandrogens, 5-alpha-reductase inhibitors, estrogens, long-acting opioid analgesics, or human growth hormone), as well as nutritional supplements that could possibly increase serum T (e.g. androstenedione or dehydroepiandrosterone).
Primary and secondary efficacy parameters
The primary efficacy variable for each study was the proportion of oral TU-treated patients who achieved a 24-h T Cavg in the eugonadal range (as defined for each study based on assay of T in serum or NaF-EDTA plasma) after two opportunities for dose adjustment. In both studies, serial blood samples were drawn at approximately −30 min and 0, 2, 4, 6, 8, and 12-h after the morning and evening oral TU doses for assay of T. Current US regulatory (i.e. US Food and Drug Administration, FDA) standards dictate that at least 75% of patients must achieve this mark with a 95% lower confidence interval (CI) of ⩾65%. Secondary efficacy was based on the proportion of patients whose Cmax aligned with the following FDA targets: ⩾85% with Cmax ⩽1500 ng/dl; ⩽5% with Cmax between 1800 and 2500 ng/dl; and no patients with Cmax >2500 ng/dl.
Study design features
Both studies were open-label, randomized trials of an oral SEDDS formulation of TU [JATENZO®; TU capsules]. Key design features are summarized in Table 1. A topical T control group [i.e. T-gel (AndroGel® 1%) or T-solution (Axiron®)] served as a positive control arm for comparative safety assessments in trials I and II, respectively. However, oral TU efficacy was evaluated independent of that observed with topical T. In each study, participants in both treatment groups were provided up to two opportunities for adjustment of their T dose based on individual responses to oral TU (per protocol) or topical T (per product labeling instructions). In general, a patient’s serum/plasma T Cavg was compared with pre-specified values that would trigger no change in oral TU or topical T dose or an up or down adjustment per protocol. The dose-adjustment algorithm for trial I was based on earlier phase II studies of oral TU, whereas the dose-adjustment paradigm for trial II was based on simulation and modeling of serum pharmacokinetic (PK) data from trial I and another clinical trial of oral TU, namely CLAR-12011 [ClinicalTrials.gov identifier: NCT01765179]. Each study assessed the impact of oral TU therapy on various patient-reported outcomes [e.g. Psychosexual Daily Questionnaire (PDQ)] 14 and, in the long-term trial CLAR-09007, the effect of oral TU on body composition and bone mineral density parameters as measured by dual-emission X-ray absorptiometry.
Key design features of phase III studies.

Percentage of subjects with testosterone Cmax < 1500 ng/dl, 1800–2500 ng/dl, and >2500 ng/dl.
Pivotal trial on which efficacy was based for FDA approval.
Meals contained typical fat content and were not required to be ‘high fat’ meals.
Blood sample taken 4–6 h after morning oral TU dose.
T Cavg calculated on basis of 12 h area under the curve; these data plus concordance analyses were used to determine best single-sample timepoint after morning oral TU dose to guide real-world dose-adjustment decisions.
The PDQ 14 was used to assess sexual function and mood changes. Patients were asked to complete the questionnaire every day for 7 consecutive days before day 1 and the last study day (end of study). Each domain of the PDQ (sexual desire, enjoyment and performance, mood, and sexual activity score) was evaluated.
a.m., morning; BID, twice daily; BMD, bone mineral density; DEXA, dual-emission X-ray absorptiometry; Cavg, time-weighted average concentration; Cmax, maximum observed concentration; FDA, US Food and Drug Administration; LC/MS-MS, liquid chromatography–tandem mass spectrometry; NaF-EDTA, sodium fluoride-ethylenediaminetetraacetic acid; PDQ, Psychosexual Daily Questionnaire; p.m., evening; T, testosterone; TU, testosterone undecanoate.
Safety measures included physical examination, vital signs, fasting clinical laboratory analysis (hematology, chemistry, urinalysis), cardiovascular (CV) biomarker monitoring [high-sensitivity C-reactive protein (hs-CRP),15,16 lipoprotein-associated phospholipase A2 (Lp-PLA2), 17 lipoprotein a (Lp(a)), 18 and apolipoprotein A1 (Apo-A1) 19 in trial I only; hs-CRP is a strong independent predictor of CV disease risk and its reduction, for example, by rosuvastatin in the JUPITER trial showed a reduction in subjects with normal LDL (see Ridker et al. 16 ). Lp-PLA2 is an inflammatory enzyme secreted by macrophages and found in atherosclerotic plaque. Epidemiological studies have demonstrated that Lp-PLA2 mass or activity is an independent risk factor for coronary or cerebrovascular events. Lp(a) is a well-recognized CV disease risk factor with elevations being associated with higher CV risk. Apo-A1 is the main constituent of HDLc and is known to drop by some degree across essentially all TRT regimens], measurement of free T, dihydrotestosterone (DHT), sex-hormone binding globulin (SHBG); luteinizing hormone (LH), follicle-stimulating hormone (FSH); PSA, and the American Urological Association/International Prostate Symptom Score (AUA/I-PSS). Prostate volume was assessed in patients enrolled in trial I by transrectal prostate ultrasound. Cuff blood pressure (BP) was measured in both studies and 24 h BP and heart-rate values were acquired every 20 min through ambulatory BP monitoring (ABPM; Spacelabs, Inc, Redmond, WA, US) on the day before the baseline visit and again, 1–3 days prior to the final PK visit in the pivotal trial (trial II).
Oral TU dose-adjustment paradigms
About 4 and 8 weeks after oral TU therapy was initiated in each trial, patients could have their TU dose adjusted based on T Cavg response in relationship to prespecified T ranges that would guide any change and the magnitude of that change. In general, dose adjustments ranged from about ± 20–30% of the prior oral TU dose based on the oral TU capsule strengths available for each study. In trial II, two approaches were taken to determine the optimal time for the assessment of circulating T on the basis of a single blood sample after the morning oral TU dose to guide dosing in real-world clinical settings where T response to TRT is determined from a single blood sample. First, extensive simulation (n = 1000 simulated patient runs) and modeling of PK data was used to identify a discrete blood sampling range (e.g. 4–6 h after oral TU) that would consistently yield a T value in close agreement with the actual T Cavg based on serial PK samples. Second, concordance analysis was performed to identify the best post-dose T assay timepoint to guide any necessary dose adjustment in oral TU patients. Concordance is defined herein to describe the extent of agreement between a decision to adjust the oral TU dose (up or down) when a single circulating T concentration remains within the hypogonadal range [i.e. <252 ng/dl (9 nmol/l) for this study] or supraphysiological range [i.e. >907 ng/dl (31 nmol/l)] and the desired outcome of that decision (i.e. a circulating T level in the eugonadal range) is achieved.
Statistical methods
The proportion of oral TU-treated subjects whose 24 h total testosterone Cavg at the final PK visit (about days 90 and 105 in trials I and II, respectively) was in the eugonadal T range was calculated for all patients. In trial I, missing PK data were not imputed from earlier values, while in trial II, patients who dropped out prior to their final PK visit due to a possible treatment-related cause (such as an adverse event) were counted as treatment failures. Consequently, PK efficacy in trial II reflects the most conservative case. For patients in trial II who dropped out for other causes (e.g. site closure not related to study conduct), their testosterone Cavg was imputed using last observation carried forward (LOCF) methodology. A 95% CI for the proportion was reported, along with the estimated proportion. Oral TU was efficacious if this proportion was at least 0.75 (75%) and the lower bound of the 95% CI was at least 0.65 (65%). The proportions of oral TU subjects with testosterone Cmax over 24 h at visit 7 that were ⩽1500 ng/dl; >1800 and ⩽2500 ng/dl; and >2500 ng/dl were calculated. Similar analyses were conducted for topical T patients, but statistical comparisons were not made between groups.
Changes from baseline for the PDQ were summarized by treatment group, and overall for each subscale score. A 95% CI for the change from baseline within each treatment group was computed. Change from baseline for other clinical parameters of interest [body composition and bone mineral density (BMD)] and the difference between treatment groups for these endpoints was compared using an analysis of covariance (ANCOVA).
Results
Patient characteristics and disposition
Table 2 summarizes key patient characteristics and disposition parameters. Notable is that in both studies, at least 90% of enrolled subjects were included in the primary efficacy analyses. In addition, few patients discontinued due to adverse events and these typically were not a result of T therapy. Mean compliance on oral TU therapy in both studies was ⩾95%.
Summary of key oral TU patient demographic and disposition parameters.

Efficacy population comprised all randomized patients who had sufficient data at day 90 PK visit to calculate serum T Cavg.
Efficacy population defined as all patients who had evaluable PK profile to calculate plasma T Cavg at final PK visit.
Day 365 of oral TU dosing.
BMI, body mass index; Cavg, time-weighted average concentration; N/A, not applicable; PK, pharmacokinetics; T, testosterone; TU, testosterone undecanoate.
Primary and secondary efficacy and testosterone time–concentration profile in response to oral TU
Primary efficacy
The primary efficacy objective of both studies was to demonstrate that a mean average T concentration could be achieved in at least 75% of oral TU patients after one or two dose adjustments. As shown in Table 3, 83.6% (trial I) and 87.3% (trial II) of oral TU patients achieved circulating concentrations of T in the mid-eugonadal range and efficacy was sustained to the 12-month timepoint in the long-term study. Efficacy in the topical T arms of each study were similar in magnitude [79.0% (trial I) and 87.3% (trial II)] and not statistically different from oral TU. The Cavg serum T on days 90 and 365 in trial I were 628 ± 343 and 524 ± 215 ng/dl, respectively [Based on data generated after completion of CLAR-09007, it became apparent that the most accurate measure of circulating T for determination of PK efficacy in men dosed with oral TU necessitated that post-collection conversion of TU to T during blood samples processing be accounted/corrected for (see Ceponis et al. 12 and Lachance et al. 13 ). We now know that the serum T values in CLAR-09007 overestimated the actual circulating level of T at the time blood was drawn by about 20% on average]. However, these mean Cavg T were associated with peak T levels at day 90 that were higher than desired (Cmax targets established by the FDA for the percentage of patients that fall into these categories at any timepoint in the efficacy trial of TRT: ⩾85% of patients with Cmax <1500 ng/dl; ⩽5% of patients with Cmax >1800–2500 ng/dl and no patients with a Cmax >2500 ng/dl). In contrast, and representative of the refinement in the initial oral TU dose and associated dose titration algorithm in trial II (pivotal trial), the mean NaF-EDTA plasma T Cavg observed was 403 ± 128 ng/dl [equivalent to approximately 489 ± 155 ng/dl in serum T units; conversion of a T concentration measured in NaF-EDTA plasma to an approximate equivalent T concentration measured 6 h after oral TU in serum required multiplying the NaF-EDTA plasma T concentration by 1.214. Data from CLAR-15012 indicated that the serum T concentration at this sample time after oral TU dosing is a reasonable approximation of Cavg. The overall correction factor is the product of three independent factors: 0.999 (to account for the small amount of overestimation in the NaF-EDTA containing tube) × 1.043 (to account for the overestimation of T that would occur in the plain tube due to TU to T conversion) × 1.166 [to account for an NaF matrix effect (NaF-EDTA plasma versus serum)] on T measurement (see Ceponis et al. 12 )]. In addition, Cmax frequencies observed in oral TU subjects in CLAR-15012 were closely aligned with desired targets.
Efficacy results for oral TU patients in trials I and II.

On final PK day after up to two dose-adjustment opportunities. Cavg in eugonadal range must be achieved by ⩾75% of oral TU patients to satisfy FDA efficacy standard.
Minimum lower bound of 95% confidence interval must be ⩾65% to satisfy FDA standard.
n = 3 patients; high Cmax occurred only after a.m. oral TU dose at single site. Further investigation indicated probable sample contamination during sample handling.
a.m., morning; Cavg, time-weighted average concentration; Cmax, maximum observed concentration; FDA, US Food and Drug Administration; N/A, not applicable; PK, pharmacokinetics; T, testosterone; TU, testosterone undecanoate.
Testosterone time–concentration profiles on the efficacy determination timepoints for trials I and II are shown in Figure 2. Both profiles are consistent and indicate peak T levels are achieved about 4 h after oral TU was administered. Mean peak T concentrations were within the eugonadal range only in the pivotal trial (trial II).

Mean concentration–time profiles for total serum T in patients treated with oral TU at final PK visit in trial I, and for NaF-EDTA plasma total T in patients treated with oral TU at final PK visit in trial II.
Single-sample dose-adjustment paradigm for oral TU
PK modeling and simulation results for circulating T confirmed that dose-titration decisions based on a single blood sample taken 3–5 h or at 4 h after the morning oral TU dose was an effective means to guide dose adjusting to achieve/maintain T concentrations in the eugonadal range. As shown in Table 4, regardless of the three measures used to determine the need to adjust the oral TU dose (i.e. Cavg based on serial PK blood sampling, a single blood sample 4 h after the morning dose or a single sample taken any time between 3 h and 5 h after oral TU), efficacy was high, with 95% of patients achieving a Cavg in the eugonadal range and <5% of patients with a mean T level below normal. Concordance analyses (data not contained herein) indicate that for the first and second PK visits, total concordance was 88% and 93%, respectively, when a single blood sample for T assay was collected 4 h after the oral TU dose. When total concordance was analyzed on the basis of a single T concentration at 6 h after oral TU at the first and second PK visit, total concordance was 98% and 96%, respectively.
PK simulation results evaluating mean T concentration (C
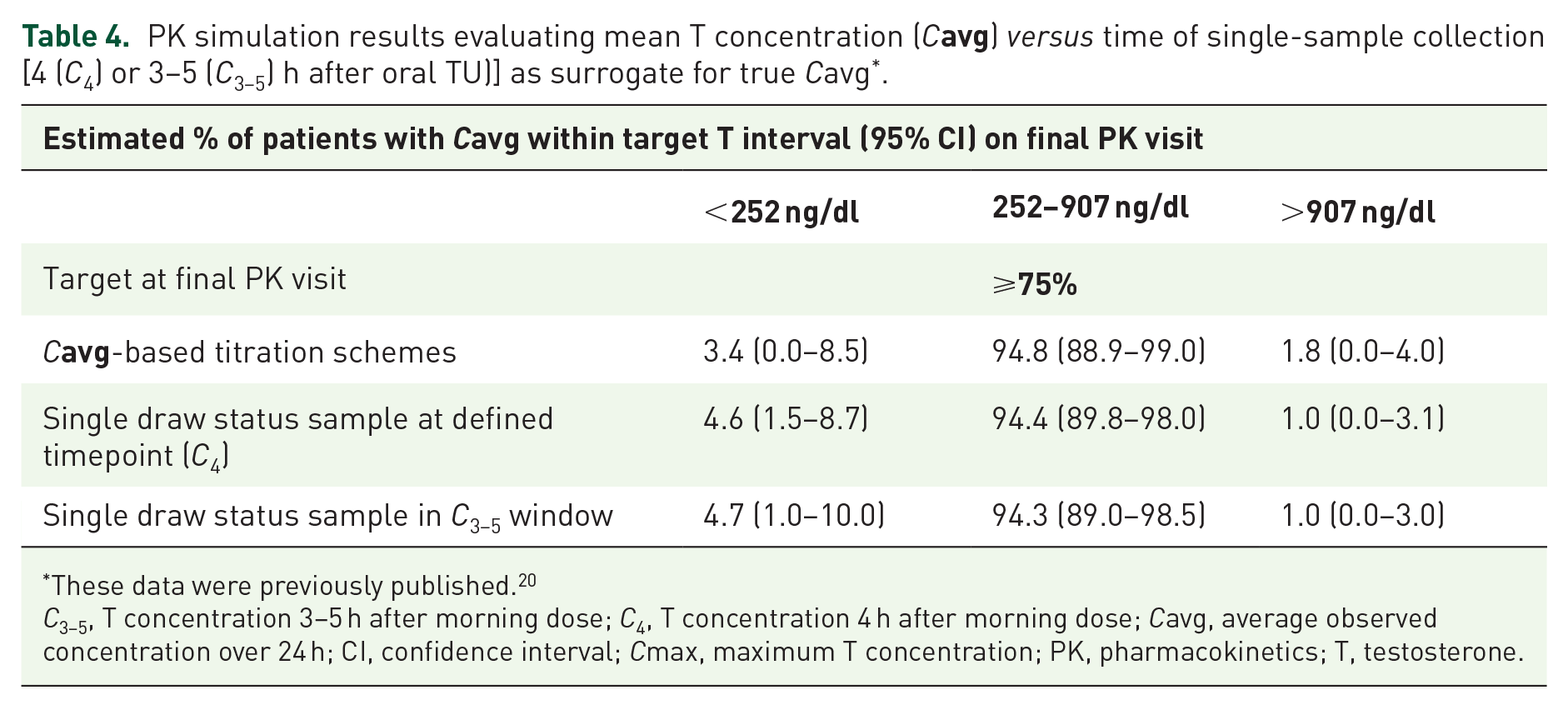
These data were previously published. 20
C3–5, T concentration 3–5 h after morning dose; C4, T concentration 4 h after morning dose; Cavg, average observed concentration over 24 h; CI, confidence interval; Cmax, maximum T concentration; PK, pharmacokinetics; T, testosterone.
Secondary efficacy
In trial I, three secondary efficacy parameters were assessed: PDQ (a measure of psychosexual function), body composition (i.e. lean and fat mass) and BMD of hip and spine. Although there was no statistically significant difference in PDQ responses between the oral TU groups, oral TU was associated with a statistically significant improvement from baseline (p <0.0001) in all PDQ responses (Figure 3). Similarly, there were no significant differences between oral TU and T-gel for spine BMD (p = 0.6002) but there were statistically significant differences between oral TU and topical T in other parameters, with the oral TU group having a greater increase in hip bone density (p = 0.0019) and lean mass (p <0.0001), and greater loss of fat mass (p = 0.0056) compared with T-gel. These differences were not only statistically significant, but clinically meaningful. During the course of the study, lean mass increased by a mean of 3.16 ± 2.70 kg from baseline, and fat mass decreased by 2.40 ± 3.64 kg at day 365 (Figure 4), both significant with a p <0.0001.

Effect of oral TU on change from baseline in psychosexual function (PDQ) responses over the 4-month treatment period in trial I.

Effect of 6- and 12-month oral TU therapy on mean (±) changes from baseline in lean body mass and fat mass in trial I.
Consistent with the findings observed in the long-term study, patients in study trial II also exhibited statistically significant improvements from baseline (p <0.0001) in all PDQ parameters shown in Figure 3. Difference in PDQ response between treatment groups was not significant.
Safety: long-term study (trial I)
No deaths occurred in response to daily oral TU exposure for 365 days. Two subjects in the oral TU group experienced an acute myocardial infarction; one in the setting of hospitalization for pneumonia. Neither event was considered to be causally related to oral TU in light of their respective cardiovascular (CV) disease histories. Treatment-emergent adverse events (TEAEs) considered by study investigators to be likely related to oral TU and T-gel exposure are summarized in Table 5. Not surprisingly, there was a higher incidence of gastrointestinal (GI) side effects associated with oral TU than topical T-gel. However, these GI effects were mild in nature and did not result in discontinuation of oral TU. Other adverse events are typical of TRT and to the extent the incidence was higher in the oral TU versus T-gel group reflected differences in delivery route or T exposure (T Cavg in oral TU group was significantly greater than in the T-gel arm).
Incidence of treatment-emergent adverse events related to long-term oral TU and T-gel therapy in study trial I.

PSA, prostate-specific antigen; TU, testosterone undecanoate.
The mean baseline hematocrit for oral TU patients was 44.1 ± 2.5%, and absolute changes after 4 and 12 months of treatment with oral TU were +2.1 ± 3.4% and +2.9 ± 3.9%, respectively, with both of these being statistically greater (p <0.0001) than the increased observed in T-gel patients. Liver function was assessed by measurement of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), total bilirubin, and alkaline phosphatase. No clinically significant changes in these parameters were observed in oral TU or T-gel patients over the 12-month course of the study, although a single patient in the oral TU group experienced two transient episodes of elevated ALT and AST that were two to three times the upper normal limit. During this period, bilirubin concentrations remained normal. At day 90, high-density lipoprotein cholesterol (HDLc) decreased more in the oral TU group (median decrease of 23.5%) compared with a decrease of 12.5% in the T-gel arm. A similar significant difference (p <0.0001) between groups was observed at day 365. No clinically significant changes over time (i.e. >5%) in oral TU patients or differences between groups was observed for low-density lipoprotein cholesterol (LDLc) or triglycerides. The effect of oral TU and T-gel on estradiol, FSH, and LH were similar. In contrast, mean free T concentrations increased from mean baseline values of 3.07 ± 1.85 ng/dl (oral TU) and 3.21 ± 1.88 ng/dl (T-gel) to values that were significantly higher in oral TU patients compared with T-gel patients at day 90 [13.37 ± 7.89 ng/dl (oral TU); 7.91 ± 4.68 ng/dl (T-gel)] and at day 365 [12.42 ± 5.81 ng/dl (oral TU); 7.62 ± 3.91 ng/dl (T-gel)]. This difference reflected a significantly greater reduction in mean SHBG in oral TU patients (−48%; days 90 and 365) compared with those treated with T-gel (−10%; days 90 and 365).
Prostate safety
We assessed prostate volume, PSA, and change in AUA/I-PSS category classification over the course of the study. Mean baseline prostate volume for oral TU was 29.3 ± 14.20 cc which was similar to the transdermal T-gel group mean baseline prostate volume of 30.7 ± 25.52 cc. Mean increase from baseline on day 365 was similar between oral TU and T-gel (2.97 ± 9.83 cc and 1.81 ± 26.40 cc, respectively) and not significantly different (p = 0.6664). At day 365, the absolute median increase for oral TU subjects was 0.150 ng/ml compared with the T-gel group increase of 0.100 ng/ml. These values were well below the clinically meaningful change of >1.4 ng/ml, within the normal range and not significantly different (p = 0.1193). Lastly, oral TU treatment did not change the AUA/I-PSS category classification, nor did it have a substantial effect on the score. Mean decrease in score from baseline at day 90 was similar between oral TU and T-gel (0.1 ± 3.9 and 0.4 ± 3.6, respectively), a finding that held consistent to day 365. Differences between the oral TU and T-gel group were not statistically significant (p = 0.3565).
Cardiovascular biomarkers: hs-CRP, Lp-PLA2, Lp(a) and Apo-A1
The impact of oral TU therapy on CV biomarkers is summarized in Table 6. For simplicity, only baseline and day 365 data are included in this table although all biomarkers were also measured on days 90 and 180. Statistical analyses compared the absolute change in baseline over time between treatment groups using a repeated-measures analysis of variance. No statistically significant difference was observed for hs-CRP [with or without exclusion of values >10 mg/l (indicative of acute infection)] or Lp-PLA2. There was a statistically significant difference in Lp(a) response in favor or oral TU [i.e. reduced mean Lp(a) versus small increase in T-gel group]. Conversely, there was a significantly greater reduction in mean Apo-A1 levels in oral TU-treated patients at day 365 (−18%) compared with the T-gel group (−6%) over the course of the study. Changes in Apo-A1 paralleled those observed in HDLc and were expected, since Apo-A1 is the main constituent of HDLc.
Effects of long-term oral TU therapy on CV biomarkers
1

1 For simplicity, only baseline and day 365 data shown. Biomarkers also assessed on days 90 and 180.
2 Analysis after excluding all values >10 mg/l. Note: analysis with all values did not affect outcome (p = 0.2781).
3 Comparison of absolute change from baseline between treatment groups based on repeated (over all assay times) measures ANOVA model.
ANOVA, analysis of variance; CV, cardiovascular; hs-CRP, high-sensitivity C-reactive protein; Lp-PLA2, lipoprotein-associated phospholipase A2; TU, testosterone undecanoate.
Finally, mean systolic BP increased slightly over the course of the study in both treatment groups, such that by day 365, the mean increase from baseline was about 5 mmHg and 3 mmHg in patients who received oral TU or T-gel, respectively. Heart rates in both groups increased about one beat per minute over the study.
Safety: trial II
No deaths occurred during the study, and there were no drug-related serious adverse events. The overall incidence of TEAEs considered related to study drug occurred in 18.7% of patients in the oral TU group and in 14.5% of the topical T group (Table 7). The proportion of patients who prematurely discontinued from the study due to adverse events was 1.8% in each treatment group.
Treatment-emergent adverse events (TEAEs) considered related to T therapy in trial II.

T, testosterone; TU, testosterone undecanoate.
The TEAEs which occurred more frequently in oral TU patients than in the topical T group were increased hematocrit, hypertension, and decreased HDLc, reported in between 3% and 5% of patients. Each of these events was reported as mild or moderate in intensity, and none resulted in premature discontinuation from the study. Decreased HDLc events occurred at the higher oral TU doses (316 mg and 396 mg BID), whereas events of increased hematocrit and hypertension were not related to TU dose nor to T Cavg or Cmax.
As expected, based on the pharmacological actions of T, mean increases from baseline in hematocrit were observed in both treatment groups at each study visit but remained within the normal range in most men (97% oral TU; 100% topical T). Shifts from normal hematocrit values at baseline to above the normal range were observed in 3% of oral TU patients at the final visit, compared with none of the topical T patients.
No clinically significant changes in the liver function tests were observed in either treatment group. Two patients on oral TU experienced an inexplicable and transient elevation in ALT and AST to levels more than twice the and upper normal limit (UNL). Bilirubin levels remained normal in these patients. A third patient experienced a transient increase of AST (also more than twice UNL) during the study. Changes in lipid profiles were more pronounced among oral TU patients compared with topical T patients. Shifts from normal baseline to below the normal range for HDLc were observed in 28.9% of oral TU patients compared with 14.8% of topical T patients at the final visit; small and clinically insignificant reductions in LDLc were observed in both oral TU and topical T patients; there was no statistically significant difference in LDLc response between groups.
Clinic systolic BP increased from baseline to the end of the study (final visit) in both treatment groups [mean ± standard deviation (SD): oral TU, 2.8 ± 11.8 mm Hg; topical T, 1.8 ± 10.8 mm Hg], whereas diastolic blood pressure was essentially unchanged at the final visit for both groups. Measurement of BP with ABPM yielded greater mean increases from baseline to end of study (approximately 2 days prior to final PK visit) in average daytime (p <0.01), night-time (p <0.01), and 24 h (p <0.002) systolic BP for the oral TU group than for the topical T group. The 24 h average systolic BP increased 4.9 ± 8.7 mm Hg in the oral TU group and 0.2 ± 9.4 mm Hg in the topical T group (p = 0.0013). Among the oral TU patients, increases in systolic BP were slightly greater in patients with a history of hypertension who were receiving antihypertensive medication (mean ± SD: 5.5 ± 8.9 mm Hg) compared with those with no history of hypertension (mean ± SD: 4.3 ± 8.6 mm Hg). There were no discontinuations of oral TU due to hypertension; 5.9% of oral TU patients initiated antihypertensive medication or required a dose increase of existing therapy. The 24 h average heart rate in the oral TU-treated group increased by two beats per minute, while in the topical T-treated group, heart rate was unchanged from baseline. The changes in heart rate were not statistically significant in either treatment group.
Discussion
The new oral TU formulation evaluated in this study becomes the first oral T-ester pro-drug approved by US regulatory authorities, and only the second oral androgen approved for TRT use in the US, the last being methyltestosterone over 60 years ago. While both trials described herein achieved primary efficacy, only trial II achieved both efficacy endpoints relative to average and peak T response. This reflects a more refined dose-titration algorithm in the pivotal versus long-term trial. We have demonstrated by two different but related analyses that a single blood sample can be collected about 4–6 h after the morning oral TU dose to assess the approximate average concentration of T over the dosing interval to reliably guide dose adjustments. In addition, concordance analyses revealed that the dose-titration algorithm employed in trial II resulted in correct clinical outcome decisions (i.e. patients achieved eugonadal T concentrations) >95% of the time. This not only validates the method used to adjust dose but should provide clinicians using this new oral TU formulation confidence that they can effectively tailor the oral TU dose for each patient. We know of no other TRT product (oral or non-oral) for which this type of concordance analysis has been conducted.
The overall safety profile of oral TU was similar in both studies and reflected the well-recognized adverse-effect profile of T therapy as a class (e.g. decreased HDLc, increased hematocrit). A minor exception to this was the occurrence of a greater number of GI-associated side effects in oral TU patients (e.g. nausea, diarrhea, burping) compared with topical T, but these were transient, minor in severity and did not result in patients discontinuing oral TU. Of particular importance is the fact that oral TU was not associated with liver toxicity in either the long- or short-term study, a sharp contrast to methyltestosterone that has been historically associated with potentially serious hepatoxicity.
Oral TU was associated with a small but statistically significant increase in systolic BP versus the topical T. This observation indicates the need for regular monitoring of BP in men receiving TRT, particularly in those with existing hypertension. The observed increase in systolic BP in some oral TU patients is consistent with effects reported for a new parenteral (subcutaneous) form of T-enanthate now marketed in the US, 21 an older formulation of TU available outside the US (Andriol®; 22 ) and an oral TU product in late-stage clinical development [ClinicalTrials.gov identifier: NCT No. 03868059]. 23 These findings indicate that the increase in BP may be a T-mediated effect and not one necessarily unique to TU or a particular route of TRT delivery. Furthermore, older studies of TRT have not looked closely nor used sensitive measurements of BP (e.g. with ABPM) that regulatory authorities now view as important for current-day evaluation of TRT safety. In an effort to determine the etiology of elevated BP in oral TU patients, we explored the potential relationship between rise in BP with numerous other factors (data not included herein), including: oral TU dose; total and free T; estradiol and DHT concentrations; and changes in hematocrit, hemoglobin (as a surrogate for viscosity and increase in plasma volume), potassium (as a surrogate of possible increases in mineralocorticoid levels/activity), and heart rate (as a surrogate of increases in beta-adrenergic receptor activity). None of these factors correlated with elevation in systolic BP. TRT is known to promote sodium and fluid retention, 24 and this may be at least one mechanism leading to elevated BP in some men dosed with oral TU. Regardless of the etiology, careful BP monitoring should be added to the other routine monitoring of men who are receiving oral TU. In addition, men treated with oral TU who have controlled hypertension should be monitored for potential increases in BP that would warrant changes to their hypertension treatment, including cessation of oral TU.
In contrast to the potential CV risk associated with elevated BP in patients treated with oral TU, we did not observe changes in other well-recognized CV risk biomarkers, namely, hs-CRP, Lp- PLA2, and Lp(a). And while we did observe a reduction in HDLc, this is a well-known effect of androgen therapy regardless of route of administration and thus oral TU therapy is not unique in producing this effect. Moreover, HDLc biology is complicated and data now suggest that its protective role in CV disease may be less important than the CV risk posed by increases in other lipid fractions, namely, LDLc and triglycerides.25,26 Thus, of greater clinical importance than reduced HDLc concentrations in oral TU patients was the lack of clinically meaningful changes in LDLc and triglyceride levels, since significant elevations in these lipid fractions (particularly LDLc) are unequivocal risk factors for CV disease.27,28
In conclusion, the new oral TU formulation described herein is a safe and effective means to treat hypogonadal men and has an overall profile consistent with the class of available TRT products. As such, this product represents a significant therapeutic advance for the treatment of appropriate hypogonadal men, particularly those in the US where, until now, an oral T treatment option was essentially unavailable. Oral TU administration is convenient, and twice-daily dosing with food (i.e. with breakfast and dinner and without the need for a high fat content) is a simple regimen that should promote better patient adherence over transdermal and injectable T products that dominate use among hypogonadal men but are associated with pain of administration (injected T-esters) or with transfer of T to women and children.
Footnotes
Acknowledgements
The authors gratefully acknowledge the investigators who participated in the conduct of trials I and II, most notably: Christina Wang, MD (Torrance, CA), John Amory, MD (Seattle, WA), Laurence Belkoff, DO (Cynwyd, PA); Gregg Flippo, MD (Birmingham, AL), Joe Blumenau, MD (Dallas, TX), Stephen Kulback, MD (Birmingham, AL), Marc Gittelman, MD (Aventura, FL), Jed Kaminetsky, MD (New York, NY), Stanon Honig, MD (New Haven, CT), Michael Zitzmann, MD (Munster, Germany), and Herman Behre, MD (Halle, Germany). We also acknowledge James Longstreth, PhD (Longstreth & Associates) and Nastya Kassir, PhD (Pharsight Consulting Services, Division of Cetara) for their PK modeling and simulation expertise; Janet Wittes, PhD (Statistics Collaborative, Inc) and Nestor Rohowsky, MS (IDCS, Inc.) for statistical analyses; Sharon Kraus (Kraus Studios) and Esther See for assistance with figures; and Theo Danoff, MD, PhD and Sandy Faulkner, RN (both Clarus Therapeutics, Inc.) for their oversight, data analyses and report preparation for trials I and II.
Conflict of interest statement
RSS served as principal investigator for studies CLAR-09007 (trial I) and CLAR-15012 (trial II) that were funded by Clarus Therapeutics, Inc. RSS has also received support from the NIH for testosterone studies in older men, and currently receives support from AbbVie as a study site for the Traverse Study of testosterone gel. He has also received support from NIH and NICHD for studies on male contraception where testosterone was a component of the experimental therapy. RED is employed by Clarus Therapeutics, Inc. (a private firm) and has an equity stake in the company.
Ethics statement
The studies described herein were conducted in compliance with all applicable country requirements for the conduct of clinical trials, including those outlined by the International Conference on Harmonization, Consolidated Guidelines on Good Clinical Practices, and the US Food and Drug Administration. Furthermore, both trials were approved by independent institutional review boards with jurisdiction over the respective study sites. Prior to participation in the trials, all patients signed informed consent documents.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.