
Abstract
Spinocerebellar ataxia type 3 (SCA3) is an inherited neurodegenerative disorder. Some of its clinical features resemble those of primary Parkinson’s disease (PD), which can easily lead to misdiagnosis. There is currently no disease-modifying therapy available for SCA3, treatment is mainly symptomatic. Herein, we report a case of a young female patient with SCA3 who presented with Parkinsonian as the main manifestation and underwent globus pallidus internus (GPi) deep brain stimulation (DBS). This is a 36-year-old female patient. Her first symptoms occurred at the age of 28 in 2009, manifesting as gait abnormalities in the right lower limb. She was misdiagnosed with early-onset PD in 2011. Genetic testing showed abnormal numbers of CAG repeats (15/70) within the coding region of the ATXN3 genes. She was diagnosed with SCA3. The patient initially responded well to levodopa-based medication, but the treatment effects gradually attenuated over time, with the development of severe symptom fluctuations and dyskinesia in 2018. The patient underwent GPi-DBS surgery in the absence of cerebellar signs, cognitive, and mood disorders. Six-year postoperative follow-up results suggest that long-term GPi-DBS is effective for the control of dyskinesia, but the residual motor symptoms (parkinsonism and ataxia) had progressively worsened in the patient. Various targets have been reported to be selected for DBS treatment of SCA3, with substantial individual differences in treatment outcomes. This case emphasizes the importance of genetic testing for the diagnosis of SCA3 and provides a basis for personalized treatment of patients with SCA3.
Keywords
Introduction
Spinocerebellar ataxias (SCAs) are a genetically heterogeneous group of autosomal dominantly inherited progressive disorders consisting of more than 40 different subtypes. 1 Among these subtypes of SCAs, spinocerebellar ataxia type 3 (SCA3) is the most common type, which is caused by an abnormal expansion of CAG trinucleotide repeats in exon 10 of the ATXN3 gene on chromosome 14. 2 The ATXN3 gene encodes the ataxin-3 protein. Affected individuals have alleles with >52 CAG trinucleotide repeats. 3 SCA3 most commonly involves the substantia nigra and cerebellar dentate nucleus (DN), leading to parkinsonian and ataxic phenotypes. 4 In rare cases of type IV SCA3, pure parkinsonism is the only symptom. There is currently no effective treatment, and variable responses to levodopa-based medicines have been reported,5–8 with the occurrence of motor/non-motor complications.9,10 Some studies have attempted to use deep brain stimulation (DBS) in two different targets including bilateral subthalamic nucleus (STN) and DN to treat SCA3, but mixed results have been reported.11,12
Herein, we report a rare case of a young woman with SCA3 who presented with levodopa-responsive parkinsonism as the first symptom and underwent DBS surgery targeting the bilateral globus pallidus internus (GPi). We describe 6-year outcomes of bilateral GPi-DBS in the patient and present an overview of all reported cases of SCA3 receiving DBS surgery, hoping to provide a basis for subsequent studies of DBS strategies in the treatment of SCA3.
Case presentation
The patient is a 36-year-old female who first presented to the Department of Neurology of our hospital in June 2020 with an 11-year history of abnormal posture and progressive bradykinesia. Her first symptoms started at the age of 28 years in August 2009, manifesting as gait abnormalities in the right lower limb while walking, foot sprains, toe walking, and uphill walking difficulties. These symptoms were particularly apparent in the morning. She was initially diagnosed with lumbar disc herniation. In 2011, the patient exhibited reduced bilateral arm swing, with the reduction more pronounced on the right side. Mild bradykinesia and rigidity were observed in the right upper and lower limbs, without resting tremor. Gait abnormalities included reduced stride length and right-sided toe walking. Postural stability was preserved, and no cerebellar signs were noted. Patient’s cognitive function, speech, and ocular movements were all normal. The patient was considered to have early-onset PD. The Hoehn–Yahr (H-Y) stage was 2. She had a Unified Parkinson’s Disease Rating Scale Part III (UPDRS-III) score of 27 and a score of 3 on the scale for the assessment and rating of ataxia (SARA). She was given 1/4 tablet of levodopa/benserazide twice a day, the treatment effect was not significant. Then the dose of levodopa/benserazide was increased to 1/2 tablet three times daily. After 1 week of treatment, the patient exhibited significant improvement and returned to normal functioning. The levodopa-equivalent daily dose (LEDD) was 300 mg/day. After treatment, the patient underwent genetic testing, which revealed abnormal numbers of CAG repeats (70, normal range: 12–40) within the coding region of the ATXN3 gene. The patient was then clinically considered to have SCA3. In 2013, nonmotor symptoms (i.e., constipation) emerged, followed by the occurrence of restless legs syndrome and motor fluctuations (i.e., “wearing-off”) in 2015. Brain dopamine transporters-positron emission tomography (DAT-PET) imaging of the patient revealed reduced distribution of 11C-CFT in the bilateral putamen and body of caudate nucleus, and the reduction was more pronounced on the left side (Figure 1). In 2016, the patient’s UPDRS-III score worsened to 39 in the “off” state and 19 in “on” state, her SARA score increased to 8, suggesting early cerebellar involvement. In 2018, she developed severe peak-dose dyskinesia and experienced shortened levodopa responses, lasting only 1–3 h. Her UPDRS-IV score rose to 3, and the LEDD reached 712 mg/day. Despite optimized medication (administration of entacapone, controlled-release carbidopa-levodopa), the patient experienced deterioration of symptoms. Brain MRI showed atrophy in the bilateral middle cerebellar peduncle and cerebellum (Figure 2). Prior to the DBS surgery in 2019, the patient underwent a levodopa challenge test with 250 mg of levodopa/benserazide, which revealed a UPDRS-III score of 43 in the “off” state and 25 in the “on” state. This demonstrates a maximum improvement rate of 41.9%, exceeding the 30% threshold for clinical significance. The UPDRS-IV score for dyskinesia was 6 during the peak dose, indicating severe “on” state dyskinesia. This condition notably impaired her quality of life and resulted in frequent falls. After obtaining informed consent from the patient and her family, bilateral GPi-DBS was performed. After DBS surgery, dyskinesia disappeared completely. However, the core motor symptoms (bradykinesia, rigidity) and cerebellar ataxia persisted and continued to progress, representing significant residual motor symptoms inadequately controlled by the combination of DBS and medication. The effects of levodopa/benserazide lasted for about 2 h, and the patient was able to take care of herself during the “on” state. At 1 year after DBS in 2020, the patient experienced difficulties with walking and generating accurate steps. Her UPDRS-III score was 62 in the “off” state and 41 in the “on” state. The H-Y stage was 3 in both “off” and “on” states, the SARA score was 22, and the LEDD was 774 mg/day. The effects of levodopa/benserazide occurred in half an hour and lasted less than 2 h, and she had difficulties in taking care of herself (Supplemental Video 1).

Cranial DAT-PET reveals decreased radioactivity distribution in the bilateral putamina and body of the caudate nuclei.

Cranial MRI of the patient. (a) Coronal view showing middle cerebellar peduncle atrophy. (b) Coronal view showing bilateral cerebellar atrophy. (c) Sagittal view showing cerebellar atrophy.
In terms of family history, the patient’s mother, second eldest sister, aunt, and grandmother had ataxia. Her second eldest sister initially developed ataxia at the age of 37 years, presented with difficulty walking and ataxic gait for 8 years (Supplemental Video 2), and developed strabismus 6 years ago. Treatment with levodopa/benserazide (1/2 tablet, three times daily) and pramipexole (0.25 mg, three times daily) was ineffective. Her second eldest sister presented 72 CAG repeats in the coding region of the ATXN3 gene. The son of her second eldest sister presented 71 CAG repeats, and he only developed strabismus, without the presence of ataxia symptoms. The patient’s eldest sister showed no abnormalities. The family pedigree is shown in Figure 3.

Family pedigree shows that the patient (the proband), her second eldest sister, mother, aunt, and grandmother all had ataxia. The son of the proband’s second sister (born in 1995) developed strabismus, but did not exhibit symptoms of ataxia.
Follow-up of the patient was performed by an independent neurologist to make necessary medication adjustments. During the postoperative follow-up period, the patient experienced progressive worsening of her condition (Supplemental Video 3). In 2021, she became wheelchair-dependent, with a UPDRS-III score of 54 in the “off” state. The LEDD was 800 mg/day. In 2022, her LEDD was 1200 mg/day. In 2024, her “on” periods lasted ⩽1.5 h, and she was bedridden (UPDRS-III score; 72 (“off” state); H–Y stage: 5; LEDD: 1499.75 mg/day). As of February of 2025, at a follow-up period of 6 years, the patient did not experience any further recurrence of dyskinesia (Figure 4). The adjustment of DBS parameters and levodopa dose over the 6-year period is shown in Table 1. An increase in levodopa dose post-DBS was noted, suggesting disease progression and progressively worsening of residual motor symptoms.
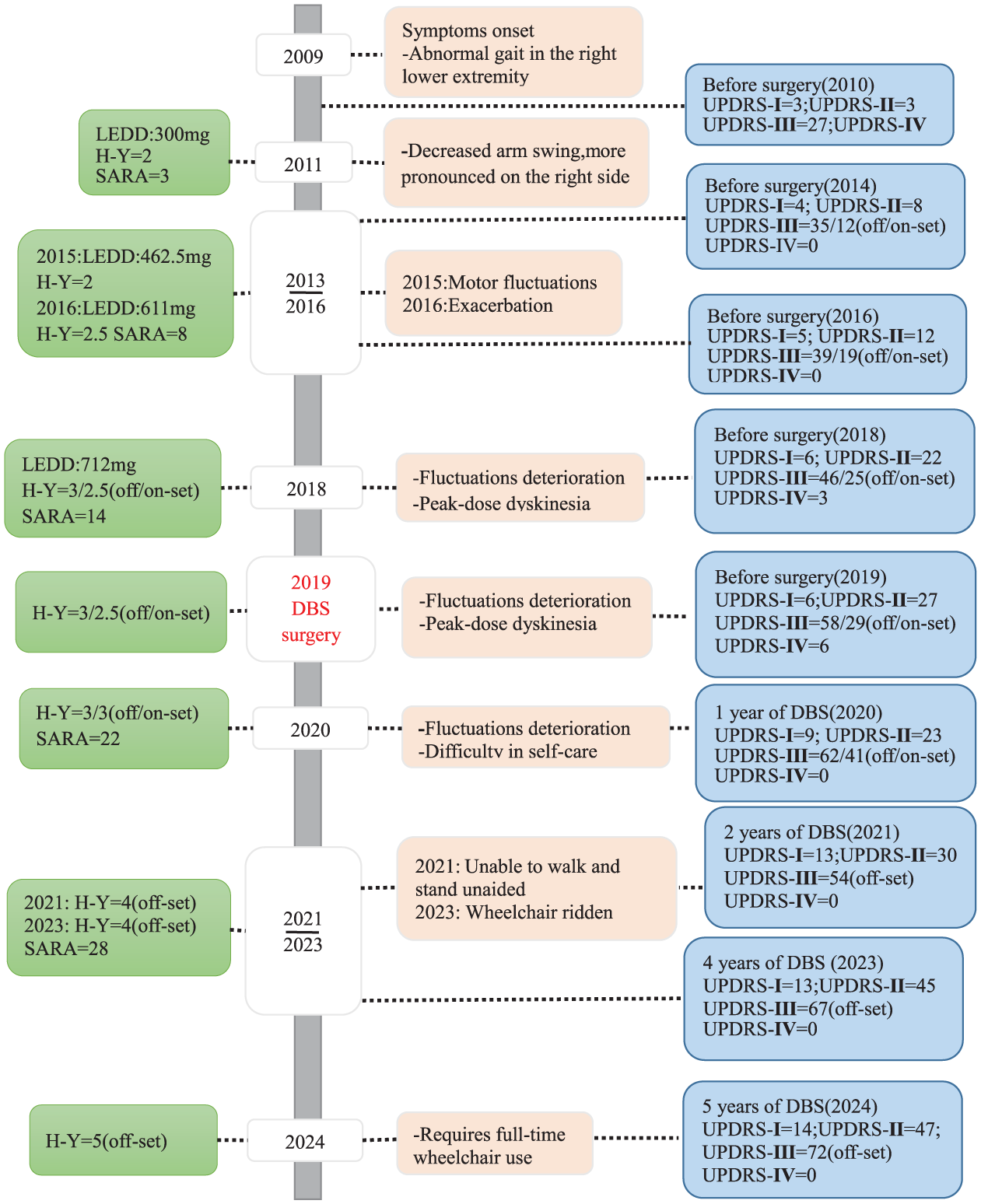
Patient’s disease course and outcomes following DBS surgery. All the reported MDS-UPDRS scores refer to “on” and “off” states. Since 2020, patient has basically been in an “off” state.
Preoperative and postoperative assessment of medications and stimulation setting.

L, left; LEDD, levodopa equivalent daily dose; R, right.
Through a literature review, we noticed that only nine patients with SCA3 treated with DBS have been reported in the literature (Table 2). In terms of demographic characteristics of these patients, the average age at the time of DBS surgery was 32.2 years old, and 55.6% (5/9) of these patients were females. The clinical manifestation was mainly ataxia (n = 7), accompanied by symptoms such as tremor, dystonia, and pyramidal tract impairment. Only one patient presented with pure parkinsonism. Surgical targets for DBS included bilateral DN in four patients, GPi in three patients, STN in one patient, DN + GPi in one patient, and ventralis oralis + GPi in one patient. The follow-up time ranged from 15 days to 4 years. There were significant individual differences in treatment outcomes. DN-DBS is considered to be effective in improving tremor and ataxia in patients with ataxia-dominant phenotype of SCA3, STN-DBS improves motor fluctuations in patients with predominant parkinsonism and motor complications. Moreover, GPi-DBS is effective for dyskinesia, which may have limited effect on residual motor symptoms.
Literature review of SCA3 treated with DBS.

AAD, age at DBS; AAO, age at onset; DBS, deep brain stimulation; DN, dentate nucleus; GPi, globus pallidum internus; ICD, impulse control disorders; LID, levodopa-induced dyskinesia; NA, not applicable; STN, subthalamic nucleus; UPDRS-III, Unified Parkinson’s Disease Rating Scale Part III; Vo, thalamic ventral-oralis complex.
Discussion
In this case report, we present a 36-year-old female patient with SCA3 confirmed by genetic analysis. Consistent with previous literature, the patient had early-onset parkinsonism as the first manifestation. She exhibited characteristic changes on DAT-PET, had good levodopa responsiveness, and later experienced typical motor complications. These clinical manifestations are generally indistinguishable from sporadic and familial PD,7,10 indicating the importance of genetic testing of dynamic mutation within the ATXN3 gene in the differential diagnosis of SCA3.
The patient responded well to low doses of levodopa and showed typical parkinsonism symptoms. These clinical manifestations were different from those observed in other members of the family. Her second eldest sister, mother, aunt, and grandmother all had ataxia as their primary clinical manifestation. Her second eldest sister and the son of her second eldest sister were diagnosed with SCA3 by genetic testing. Notably, the son of her second sister did not exhibit obvious ataxia symptoms except for strabismus. Based on phenotypic variation of SCA3, three clinical phenotypes have been described: type I is characterized by pyramidal and extrapyramidal symptoms, with an early age of onset; type II is characterized by cerebellar and pyramidal symptoms; and type III is characterized by cerebellar signs and anterior horn cell degenerative symptoms. 13 A rare type IV is characterized by parkinsonism and peripheral neuropathy. 2 Although parkinsonism symptoms have been occasionally reported in SCA3 patients, levodopa-responsive parkinsonism as a dominant clinical phenotype has rarely been described.
This intrafamilial heterogeneity in clinical manifestations suggests that the clinical phenotype of SCA3 may be highly variable, which trend to be associated with different CAG repeat lengths in the ATXN3 gene. The CAG repeat number usually ranges from 12 to 44 in normal individuals, and 52 to 200 in patients with ataxia. Patients with parkinsonian phenotype of SCA3 tend to have fewer CAG repeats than those with ataxic phenotype of SCA3. 14 In this case report, the number of CAG repeats in the patient, her second eldest sister, and the son of her second eldest sister were 70, 72, and 71, respectively. The results are consistent with the literature. Although ATXN3 misfolding caused by abnormal polyglutamine expansion is a key player in the pathogenesis of SCA3, 15 intrafamilial phenotypic heterogeneity may also be closely related to the complex pathological processes of the disease. 16 As we reported in this case, the patient and two family members had different phenotypes of SCA3. Parkinsonism may be attributed to substantia nigra lesions and ataxia may be due to DN injury and atrophy of the dorsal nucleus of Clarke column. The clinical presentation of SCA3 is variable, reflecting the involvement of a wide range of structural lesions in the central and peripheral nervous system.17,18
In this case, GPi-DBS provided certain symptomatic benefits, particularly in the management of dyskinesia. After DBS surgery, the patient exhibited complete resolution of dyskinesia, but progressively worsening of residual motor symptoms (parkinsonism and ataxia). Both parkinsonism and ataxia can continue to progress despite DBS surgery. These clinical outcomes are consistent with previous reports in the literature. 19 To date, only a limited number of case reports have described the effects of DBS in SCA3, and the outcomes appear heterogeneous. This variability highlights the importance of phenotype-specific target selection. For instance, STN-DBS may be more suitable for patients with predominant parkinsonism and motor complications, as illustrated in one case where a patient with SCA3 showed sustained improvement in both parkinsonian symptoms and motor complications over 63 months. 11 Another study reported that STN-DBS led to marked improvement in flapping tremor at 4–7 Hz by 6 months postsurgery, but long-term follow-up data are not available. 20 While GPi-DBS is a well-established treatment for PD and refractory dystonia,21,22 its benefits in SCA3 appear limited to certain genetic subtypes.23–26 Conversely, DN-DBS may be more appropriate for ataxia-dominant phenotypes, with some reports indicating improvements in tremor and gait.12,24 However, its effects on noncerebellar symptoms, such as dystonia, remain unclear. Overall, target selection should be guided by the dominant clinical phenotype (e.g., parkinsonism vs ataxia), disease stage (early vs advanced neurodegeneration), and biomarker profiles—such as neurofilament light chain levels and MRI volumetry. 17
The mechanisms through which DBS may exert symptomatic effects in SCA3 could involve its distinct modulation of both the pallidothalamic and dentatorubrothalamic networks. The pallidothalamic pathway, which connects the GPi to the ventral anterior/ventral lateral thalamic nuclei, appears to play a key role in motor control. The improvement in dyskinesia observed in our patient after DBS may be achieved through modulation of the pallidothalamic pathway. In contrast, the dentatorubrothalamic pathway, which links the cerebellar DN to the thalamus via the red nucleus, is crucial for cerebellar coordination. The persistent ataxia despite the implementation of GPi-DBS suggests that the modulation of cerebellar outflow pathways through GPi stimulation alone is limited, highlighting the network-specific nature of DBS effects.20–22
In this patient, GPi-DBS was effective in controlling dyskinesia by modulating GPi output, but it failed to address two crucial components of the disease: the progressive nigrostriatal degeneration that underlies the residual motor symptoms and the cerebellar dysfunction that drives ataxia. Our results contrast with the findings of Kuo et al., 11 who reported better response to STN-DBS, with improved parkinsonian symptoms. In this study, the target selection prioritized dyskinesia control over other symptoms, reflecting the clinical heterogeneity in SCA3. The variable outcomes across different DBS targets (STN, GPi, DN) in literature emphasize the need for phenotype-specific surgical strategies,11,12,19,24–26 and tailoring the strategies to each patient’s predominant symptoms and underlying neurodegenerative patterns.
The limited therapeutic response observed in this SCA3 patient following GPi-DBS may be attributed to several factors: (1) Differences in DBS target selection compared to previous reports; (2) The inherent genotypic and phenotypic variability among SCA3 patients; (3) The longitudinal SARA scores in our patient highlight the relentless cerebellar decline despite the implementation of GPi-DBS, this aligns with the biomarker-defined progression of SCA3. 17 The beneficial effects of GPi-DBS on levodopa-induced dyskinesia and its limited effect on ataxia progression reflects the diffuse neurodegeneration in SCA3, particularly affecting cerebellar and pontine structures. This supports the hypothesis that the efficacy of DBS in SCA3 may be confounded by the stage and distribution of neurodegeneration at the time of intervention. Future studies should correlate DBS outcomes with biomarker profiles (e.g., NfL, MRI volumetry) to identify optimal surgical timing. The findings in this case suggest that when considering DBS for SCA3, factors such as disease stage, clinical features, and genetic background need to be taken into account to optimize target selection and timing of surgery. Additionally, levodopa dose adjustment after DBS may also influence the outcomes. Future studies with larger sample sizes and long-term follow-up is required to further clarify the indications, predictive factors, and optimal medication adjustments for efficacy of DBS in the treatment of SCA3 and provide a basis for the formulation of personalized treatment plans. Furthermore, keeping the stimulation parameters (pulse width and frequency) constant throughout the 6-year follow-up period may be considered as a limitation of the present case report, the potential impact of alternative stimulation parameters or contact configurations on progressive parkinsonian and ataxic symptoms should be explored in future studies.
This case has an important clinical guiding value, which is mainly reflected in the following three aspects: First, this case report warns the clinicians that SCA3 should be included in the differential diagnosis even if the patient exhibits typical features of early-onset PD, and genetic screening is a powerful tool for confirming the diagnosis. Second, the case report reveals a noteworthy phenomenon. The patient showed “dramatic” responses to low-dose levodopa, but later developed severe motor complications, whereas his second sister who was also diagnosed with SCA3 did not respond to levodopa. This difference suggests that there is remarkable heterogeneity in levodopa responsiveness among patients with SCA3. This may be related to different subtypes of SCA3 or individual differences in genetic background. Systematically tracking disease progression in SCA3 patients with different genetic backgrounds will help to establish prognostic prediction model that can be used before DBS and provide a scientific basis for clinicians to screen the most suitable patients for DBS surgery.
Supplemental Material
sj-pdf-1-tan-10.1177_17562864251396525 – Supplemental material for Long-term globus pallidus internus deep brain stimulation in a young patient with spinocerebellar ataxia type 3 initially presenting with levodopa-responsive parkinsonism: a 6-year follow-up case report and literature review
Supplemental material, sj-pdf-1-tan-10.1177_17562864251396525 for Long-term globus pallidus internus deep brain stimulation in a young patient with spinocerebellar ataxia type 3 initially presenting with levodopa-responsive parkinsonism: a 6-year follow-up case report and literature review by Jing Zhao, Shaochen Ma, Yunlei Gao, Jia Chen, Jiaqi Chen, Chong Shi, Peifu Wang, Jilai Li, Jichen Du and Zhirong Wan in Therapeutic Advances in Neurological Disorders
Footnotes
Acknowledgements
We gratefully appreciate all the participants and staff for their contributions.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.