
Abstract
Objective:
To report on safety and effectiveness of subcutaneous cladribine (Litak®) in multiple sclerosis (MS) patients.
Methods:
Litak® was offered to MS-patients irrespective of disease course. Litak® 10 mg was administered for 3–4 days during week 1. Based on lymphocyte count at week 4, patients received another 0–3 doses at week 5. A second course was administered 11 months later. Follow-up included adverse events, relapses, expanded disability status scale (EDSS), 9-hole-peg and Timed-25-foot-walking tests, no-evidence-of-disease-activity (NEDA), no-evidence-of-progression-or-active-disease (NEPAD), MRI, cerebrospinal fluid (CSF) neurofilament light chain (NfL), and lymphocyte counts.
Results:
In all, 208 patients received at least one course of treatment. Age at baseline was 44 (17–72) years and EDSS 0–8.5. Cladribine was generally well tolerated. One myocardial infarction, one breast cancer, and three severe skin reactions occurred without long-term sequelae. Two patients died (one pneumonia, one encephalitis). Lymphopenia grade 3 occurred in 5% and grade 4 in 0.5%. In 94 out of 116 pwMS with baseline and follow-up (BaFU) data after two treatment courses, EDSS remained stable or improved. At 18 months, 64% of patients with relapsing MS and BaFU data (n = 39) had NEDA. At 19 months, 62% of patients with progressive MS and BaFU data (n = 13) had NEPAD. Of n = 13 patients whose CSF-NfL at baseline was elevated, 77% were normalised within 12 months.
Conclusions:
Litak® was well tolerated. Effectiveness in relapsing MS appeared similar to cladribine tablets and was encouraging in progressive MS. Our data suggest cladribine may be safe and effective in MS-patients irrespective of their disease stage.
Introduction
Multiple sclerosis (MS) is a chronic immune-mediated, demyelinating, and degenerative disease of the central nervous system (CNS) affecting about 2.8 million Worldwide. 1 While the course of MS is highly variable, it is generally associated with chronic accelerated loss of CNS tissue and disability accrual.
Numerous disease-modifying treatments (DMT) have been licenced for people with MS (pwMS). However, access to DMT is highly regulated. In the United Kingdom (UK), DMT-eligibility is established by the National Institute for Health and Care Excellence (NICE), Scottish Medicines Consortium and Belfast Health and Social Care Trust.
Cladribine tablets, CladT, were first licenced in Australia in 2010. However, CladT were suspended less than 12 months after rejection of licence applications to the European Medicines Agency (EMA) and U.S. Food and Drug Administration in 2010/11. 2 Key for these rejections were safety concerns related to lymphopenia and a potential risk of malignancies. However, independent research into the mechanism of action of cladribine in MS,3–5 the cancer risk 6 and publication of further data from the pivotal studies5,7 supported a reappraisal and ultimately licencing of CladT (Mavenclad®) from August 2017. In the UK National Health Service (NHS), Mavenclad® was subsequently fast-tracked to become the first MS-DMT of the Accelerated Access Collaborative. 8
We started using subcutaneously injected cladribine (Litak®), licenced for treatment of hairy-cell leukaemia since 1993, 9 as an off-label immunotherapy from late 2014, that is, 3 years prior to EMA licencing of Mavenclad®. As part of our plans to make cladribine available to pwMS we confirmed, in a meeting with the UK Medicines and Healthcare Regulatory Authority on 18 April 2013, the bioequivalence of parenteral and oral cladribine. Dosing, however, had to be adjusted to 100% bioavailability of parenteral cladribine compared to about 42% of CladT. 10 We previously described preliminary findings in our emerging cohort of pwMS undergoing Litak® treatment. 9 Here, we report on a significantly expanded cohort including new information on the risks and potential benefits of this treatment.
Methods
This service development was registered with the Clinical Effectiveness Unit at Barts Health NHS Trust (Barts Health; registration number: 11483). The use of Litak® in pwMS was approved by the Barts Health Neuroscience Clinical Network.
From October 2014, pwMS were offered Litak® irrespective of whether they were eligible for NHS DMT. Patients considering the treatment were provided with comprehensive information to aid their understanding and decision regarding Litak® treatment. 11
Between October 2014 and February 2016, treatment decisions were largely based on clinical judgement (chronic disability accrual, relapses) although gadolinium-enhancing (Gd +) lesion(s) and new/enlarging T2 lesion(s) on MRI were considered. From February 2016 onwards disease activity based on MRI and/or cerebro-spinal fluid (CSF) neurofilament light chain (NfL) levels, over and above clinical activity, was mandatory for treatment eligibility. 12 Treatment with Litak® in all cases was approved by the neuroinflammation multidisciplinary team of Barts Health.
Safety review
Prior to treatment initiation, pwMS had to pass a safety checklist 11 including blood tests (full blood and differential white cell counts, biochemistry, liver function tests, creatinine) and urinalysis. The presence of active and/or latent infections was ruled out, and the absence of other contraindications, as per Litak® SmPC confirmed. Minimum lymphocyte count (TLC) had to be 1.0 × 109 L−1. Active malignancies were excluded based on history and clinical examination. Compliance with regular cervical cancer screening was mandatory.
In patients without immunoreactivity against Varicella zoster virus, vaccination was mandatory. We also recommended concomitant famciclovir 500 mg twice daily as a herpes prophylaxis for 60 days from the first Litak® administration. Reassured that very few cases developed significant lymphocyte toxicity, the dose of famciclovir was reduced to 250 mg per day. We discontinued antiviral prophylaxis after Mavenclad® was licenced in August 2017.
Patients of childbearing potential had to use effective contraception during treatment, and for at least 6 months after receiving the last dose of Litak®. Due to the potential impact of cladribine on male fertility as per Litak® SmPC, 13 men were offered cryopreservation of sperm.
Informed consent was documented using a form designed in 2015, revised in 2016 and 2018. 11
Treatment
The Litak® dose administered to each patient was personalised to body weight and total lymphocyte count (TLC) as previously described.9,14 In short, Litak® 10 mg was administered on three consecutive days (four if body weight was >90 kg). Based on TLC at week 4, another 0–3 injections were given in week 5 (Figure 1). The treatment course was repeated in weeks 48 and 52, again depending on weight and TLC.

Dosing schedule of Litak® adjusted to body weight and lymphocyte count. Treatment course 1: Following injections of Litak® 10 mg on 3–4 consecutive days in week 1, total lymphocyte count (TLC) was measured at week 4. If TLC was ⩾1.0 × 109 L−1, further injections of Litak® 10 mg were given on three consecutive days in week 5. If TLC was 0.8–0.9 × 109 L−1, further injections of Litak® 10 mg were given on two consecutive days in week 5. If TLC was 0.5–0.7 × 109 L−1, one further injection of Litak® 10 mg was given in week 5. If TLC was <0.5 × 109 L−1, no further injection of Litak® was given during this treatment course. Treatment course 2 A: If TLC at week 44 was ⩾1.0 × 109 L−1, injections of Litak® 10 mg were given on three consecutive days in week 48. TLC was then measured at week 51. If TLC was ⩾1.0 × 109 L−1, further injections of Litak® 10 mg were given on three consecutive days in week 52. If TLC was 0.8–0.9 × 109 L−1, further injections of Litak® 10 mg were given on two consecutive days in week 52. If TLC was 0.5–0.7 × 109 L−1, one further injection of Litak® 10 mg was given in week 52. If TLC was <0.5 × 109 L−1, no further injection of Litak® was given. Treatment course 2B: If TLC at week 44 was 0.8–0.9, injections of Litak® 10 mg were given on two consecutive days in week 48. If TLC was 0.5–0.7 × 109 L−1, one further injection of Litak® 10 mg was given in week 48. If TLC was <0.5 × 109 L−1, no further injection of Litak® was given.
Outcomes
Patients were clinically assessed at baseline and annually thereafter, though some were seen more frequently. Relapse and expanded disability status scale (EDSS) 15 data were extracted from the electronic health record at the point of care. From September 2016, we introduced the nine-hole-peg (9HPT) and Timed-25-foot-walking (T25FW) tests as additional assessments. 16 Follow-up included adverse events (AEs), presence/absence of Gd+ lesions on T1-weighted MRI and new lesion(s), or an increased lesion size, compared to a reference scan on T2-weighted MRI. CSF-NfL levels were quantified using a commercial ELISA (UmanDiagnostics NF-Light ELISA). 17 The upper limit of normal (ULN) of the CSF-NfL level was 290 pg/mL for patients aged <30 years, 380 pg/mL for 31–39 year olds, and 830 pg/mL for patients ⩾40 years. 18
Disability progression was defined as an increase in EDSS between two assessments of (1) ⩾1 point if baseline EDSS was ⩽5.5 and (2) ⩾0.5 point if baseline EDSS was ⩾6.0. Cut-offs of 20% change (speed) were used for both T25FW and 9HPT. 19 The proportion of patients with ‘no evidence of disease activity’ (NEDA) and ‘no evidence of progression or active disease’ (NEPAD) were calculated for patients with complete datasets. Since a number of patients in our cohort were unable to complete the T25FW, the test was dropped to create a measure called NEPAD∗. Figure 2 illustrates the process of selecting suitable patients with complete datasets. NEDA was defined as no evidence of disability progression measured using the EDSS, absence of relapses and of MRI activity. NEPAD was defined as no evidence of disability progression measured using EDSS, absence of progression on T25FW and 9HPT and absence of both relapses and MRI activity. NEPAD* was defined as NEPAD disregarding the T25FW.
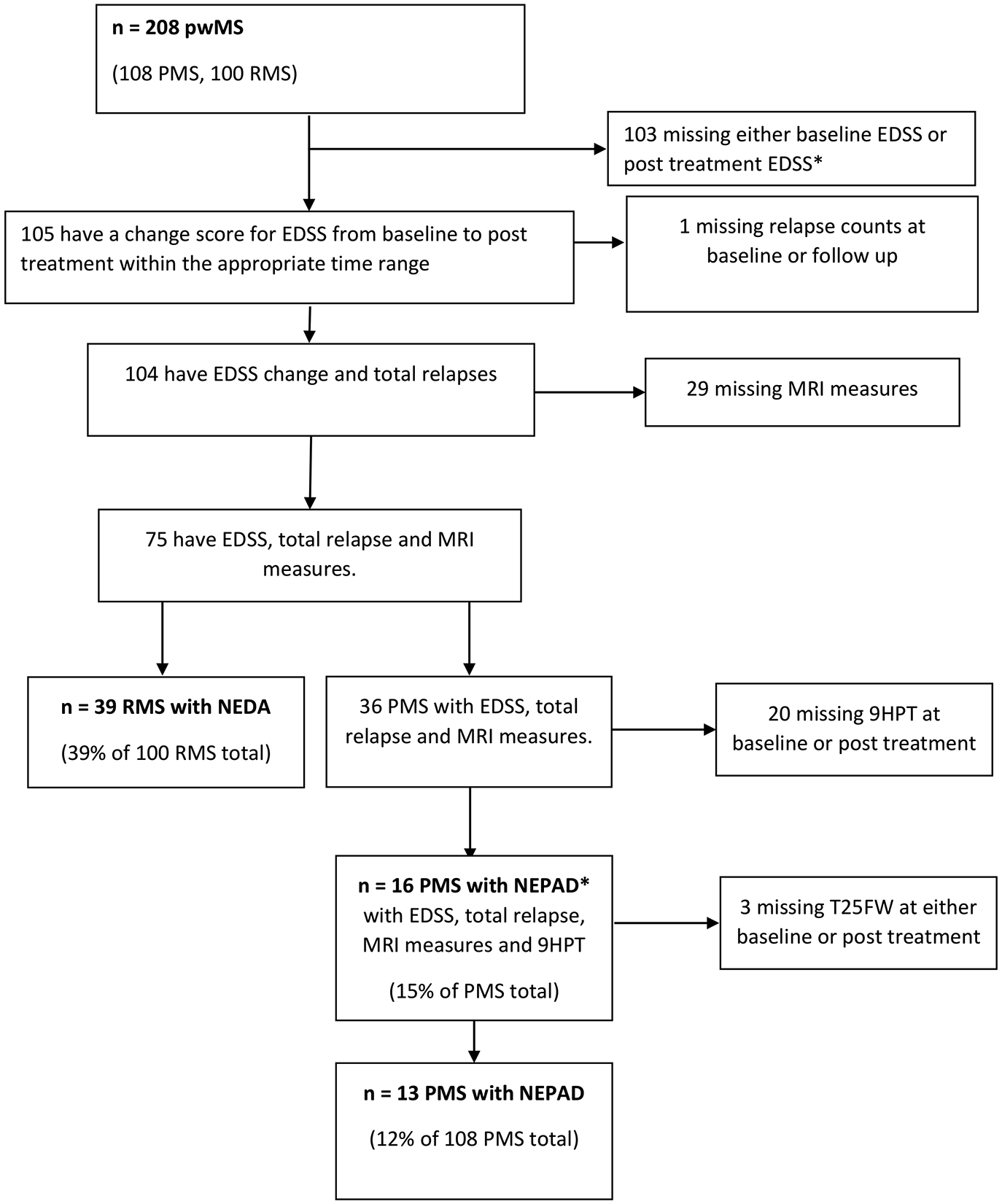
CONSORT flow diagram showing patient selection for No Evidence of Disease Activity (NEDA), No Evidence of Progression or Active Disease (NEPAD) and NEPAD less the Timed 25 foot Walking Test (NEPAD*) analysis based on criteria for data completeness.
Statistical analysis
Descriptive statistics are reported as mean/median (range) for continuous variables and as frequencies (%) for categorical variables. Safety and para-/clinical efficacy after two treatment courses was compared to baseline. The analysis was performed using Stata 16.
Results
Between October 2014 and November 2018, a total of 208 pwMS (131 females, 77 males) received at least one course of Litak® (Table 1); 196 patients (94%) completed two courses. See Table 1 for demographic and clinical characteristics. Age at baseline was 44 years (mean; range: 17–72 years). Disease duration was 11 years (mean; range: 1–48 years). One-hundred patients had relapsing (RMS) and 108 progressive (PMS) disease. Of those with PMS, 62 had SPMS and 46 PPMS. EDSS was 5.5 (median: 0–8.5). One-hundred-and-thirteen patients were eligible for NHS DMT, but chose Litak® instead. Ninety-five patients were not eligible for NHS DMT. The cohort includes seven patients previously reported as case studies.12,20–22
Baseline demographics and clinical characteristics of 208 patients with multiple sclerosis treated with Litak®. Results are shown for the whole cohort and separately for relapsing MS (RMS) and progressive MS (PMS).
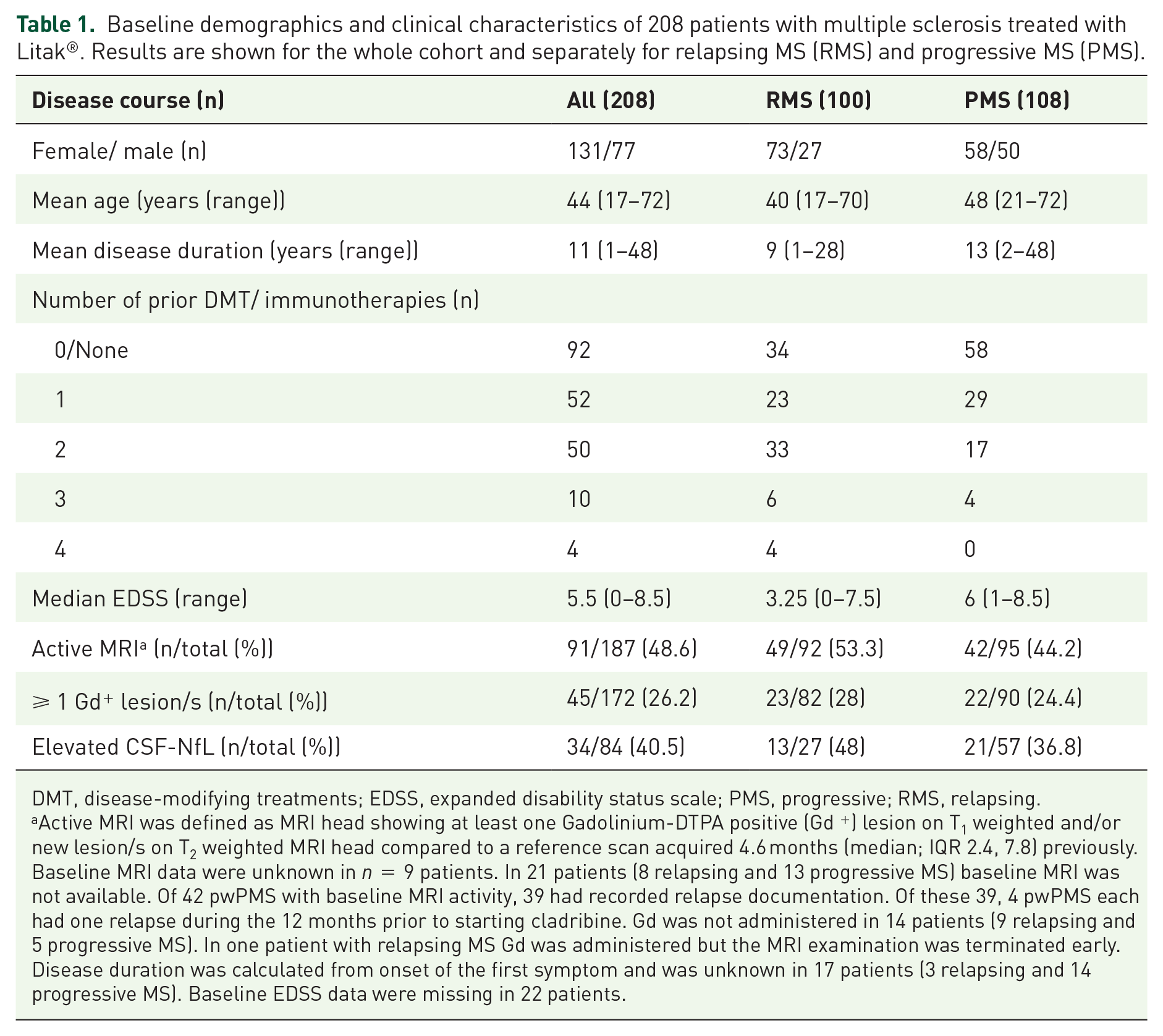
DMT, disease-modifying treatments; EDSS, expanded disability status scale; PMS, progressive; RMS, relapsing.
Active MRI was defined as MRI head showing at least one Gadolinium-DTPA positive (Gd +) lesion on T1 weighted and/or new lesion/s on T2 weighted MRI head compared to a reference scan acquired 4.6 months (median; IQR 2.4, 7.8) previously. Baseline MRI data were unknown in n = 9 patients. In 21 patients (8 relapsing and 13 progressive MS) baseline MRI was not available. Of 42 pwPMS with baseline MRI activity, 39 had recorded relapse documentation. Of these 39, 4 pwPMS each had one relapse during the 12 months prior to starting cladribine. Gd was not administered in 14 patients (9 relapsing and 5 progressive MS). In one patient with relapsing MS Gd was administered but the MRI examination was terminated early. Disease duration was calculated from onset of the first symptom and was unknown in 17 patients (3 relapsing and 14 progressive MS). Baseline EDSS data were missing in 22 patients.
Ninety-two patients (44%) were DMT naive; 116 (56%) received at least one licenced or off-label immunotherapy prior to Litak® treatment; 52 patients (26%) received one, 50 (24%) two, 10 (5%) three, and 4 (2%) four drug(s) prior to commencing Litak®. Of patients receiving DMT prior to Litak® treatment, 66 had RMS and 50 PMS. The last DMTs patients received prior to starting Litak® are listed in Table 2. The most common prior DMTs were dimethyl fumarate, fingolimod and beta interferons; 51 out of 116 (44%) patients started treatment with Litak® due to evidence of disease activity and/or clinical deterioration. This was based on MRI only in 13 (11%), relapses in 12 (10%), and clinical progression in 21 (18%); in three patients, no details of disease progression were documented. In three patients, both disease progression and MRI activity were detected. One patient had a relapse and detectable MRI activity, one switched from dimethyl fumarate due to MRI activity and problems with taking a daily tablet. One out of 116 pwMS received CladT 3.5 mg/kg over 2 years during both the CLARITY and CLARITY extension trials (total dose 7 mg/kg). 23 She then went onto interferon β-1a (Avonex®), 54 months after completing her second full treatment cycle with CladT. Due to MRI activity, she started Litak® after 7 months of Avonex® treatment.
Overview of the last immunotherapy received prior to Litak® in 116 patients with multiple sclerosis.
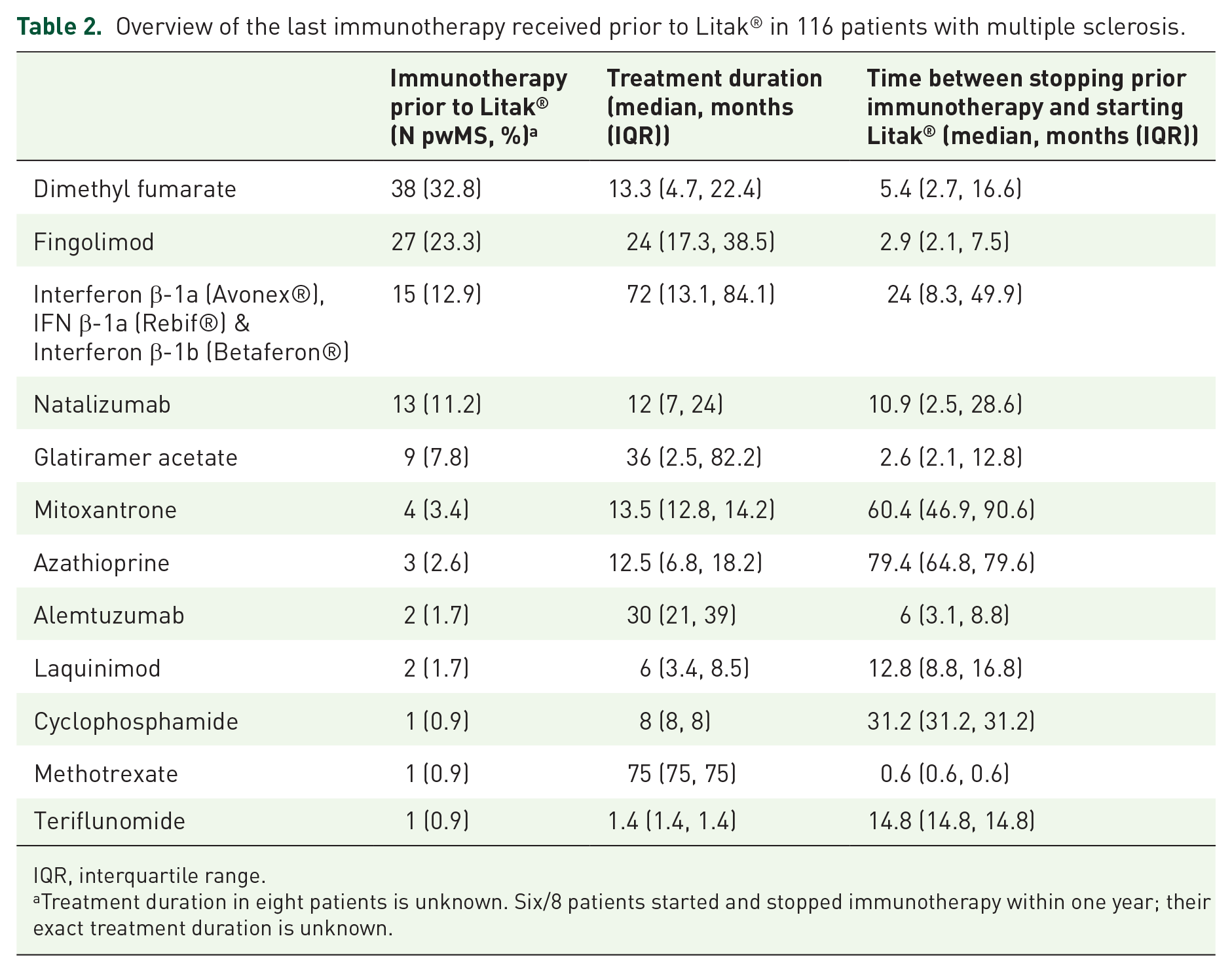
IQR, interquartile range.
Treatment duration in eight patients is unknown. Six/8 patients started and stopped immunotherapy within one year; their exact treatment duration is unknown.
In all, 65 out of 116 pwMS (56%) went on treatment with Litak® due to AEs, risk considerations of their current immunotherapy, or other reasons. Four patients had previously been treated with off-label immunotherapies (two mitoxantrone and one each cyclophosphamide and azathioprine). Six patients had been participating in clinical trials for PMS and were left without immunotherapy; two each participated in the ASCEND study of natalizumab in secondary progressive (SP) MS, 24 the INFORMS trial of fingolimod20,25 in primary progressive (PPMS) and the Arpeggio trial of laquinimod in PPMS. 26
One patient felt they had no benefit from taking dimethyl fumarate; one had been treated with interferon beta until diagnosis of SPMS and was left without immunotherapy; one was on fingolimod and considered at high risk of progressive multifocal leukoencephalopathy (PML); one had extended travel plans and switched within 4 weeks from natalizumab to Litak® to avoid monthly infusions. Two patients developed anti-natalizumab antibodies. In two cases, no reasons were documented.
The reasons why 12 pwMS (6%) did not receive a second course of Litak® included personal choice in five and one each of difficulty travelling to our centre, switch to haematopoietic stem cell transplantation (HSCT) and switch to fingolimod due to relapse. Two of 208 patients deferred their second course due to planned pregnancy. One patient died prior to receiving their second course. One patient did not receive their second course due to ongoing lymphopenia (0.4 × 109 L−1). Notably, this patient underwent chemotherapy for Hodgkin’s lymphoma 28 years prior to starting Litak®. Moreover, Litak® was preceded by dimethyl fumarate treatment for 22 months, which was associated with lymphopenia. Her most recent TLC, 37 months after her single course of Litak®, was 0.9 × 109 L−1.
Of the 187 patients who had an MRI-head prior to their first cladribine injection, 91 (49%) displayed activity (Table 1).
Since our dosing schedule was adapted to both body weight and individual total lymphocyte count (TLC), only 75% received the maximum dose (60–70 mg) at their first course while 25% received a reduced dose based on TLC (Figure 1). Of 208 patients treated, 28 (14%) weighed >90 kg and thus received 40 mg (rather than 30 mg in pwMS ⩽90 kg) during week 1 of their first treatment course. Of these 28 patients, 18 received 70 mg in total during their first course. Of 196 pwMS receiving a second course of Litak®, 35% received the full dose (60–70mg) while in 65% the dose was reduced based on their TLC (Figures 3A and 3B).

(a) Proportion of patients receiving the maximum and reduced dose of Litak® at courses 1 and 2. (b) Total dose of Litak® administered at courses 1 and 2.
Safety and tolerability
See Table 3 for a summary of AEs. Overall, Litak® was well tolerated.
Overview of adverse events in a cohort of 208 patients with multiple sclerosis.

CNS, central nervous system; MS, multiple sclerosis.
However, two patients died. The first patient was admitted to a different hospital. He was 61 years old and diagnosed with MS 16 years before starting Litak®. He died with an EDSS of 6.5, 44 weeks after completing his second course of Litak®. The death certificate stated ‘encephalitis’. No microorganism was identified and no autopsy performed.
The second patient died from H1N1 influenza-associated pneumonia, as reported previously. 22 This 53-year-old woman had an EDSS of 8.5 and died 11 weeks after completing her first course of Litak®. Of note, her TLC was within normal range (1.1 × 109 L−1) 4 weeks after completing the course, but was then admitted at the beginning of week 11 with pneumonia and grade 4 lymphopenia 22 ; she died shortly after.
One case of grade 3 invasive ductal carcinoma/high-grade ductal carcinoma in situ occurred (lymph node negative/HER2-negative/ER-positive), 10 months after completing the second course of Litak®. Breast lumpectomy followed by radiotherapy was undertaken. The patient’s oncotype Recurrence Score was 23. No chemotherapy was given. She has fully recovered with no evidence of malignancy on two annual follow-up mammograms and remains on treatment with tamoxifen.
As reported previously,9,21 one patient suffered a myocardial infarction 6 weeks after starting Litak® and three developed a severe allergic skin reaction (SASR) with complete resolution within weeks on steroids and antihistamines.
Lymphocyte kinetics
Of 208, 34 (16%) did not have any lymphopenia in the course of Litak® treatment (Figure 4). Grades 1 and 2 lymphopenia occurred in 49 (24%) and 113 (54%), respectively. Grade 3 lymphopenia occurred in 11 (5%): 3 patients at weeks 4, 2 patients at week 20, and in one patient each at weeks 32, 44, 51, 56, 68, and 92. Grade 4 lymphopenia was detected in the patient described above who died of influenza-associated pneumonia.

Total lymphocyte count (TLC) in patients treated with Litak® dose-adapted to body weight and TLC.
Clinical effectiveness
The clinical outcomes are summarised in Table 4 and Figure 5. Median EDSS after a mean follow-up of 17 months increased from 5.0 to 5.5. Of 116 patients where EDSS scores were available at two time points, 94 (81%) were stable or improved, while 22 out of 116 (19%) deteriorated. In 130 patients where relapse data were available at two time points, eight patients (6%) relapsed. The mean MRI follow-up period was 19 months. In 121 of 147 patients (82%) with MRI head data at two time points available, no T2 or Gd+ lesion activity was detected at follow-up.
Clinical outcomes in patients with multiple sclerosis treated with Litak®.

EDSS, expanded disability status scale; 9HPT, Nine-hole-peg-test; T25FW, Timed 25 foot walking-test, MRI, Magnetic resonance imaging; PMS, progressive multiple sclerosis; RMS, relapsing multiple sclerosis.
Outcomes at baseline and follow-up for patients with data at both timepoints available. EDSS and relapse follow-up period was 17 months (mean; range 14–21); MRI follow-up period of 19 months (mean; range: 16–23). Nine-hole-peg test follow-up period was 13 months (mean; range 12–14). Timed-25-foot-walking test follow-up period was 13 months (mean; range: 12–16).
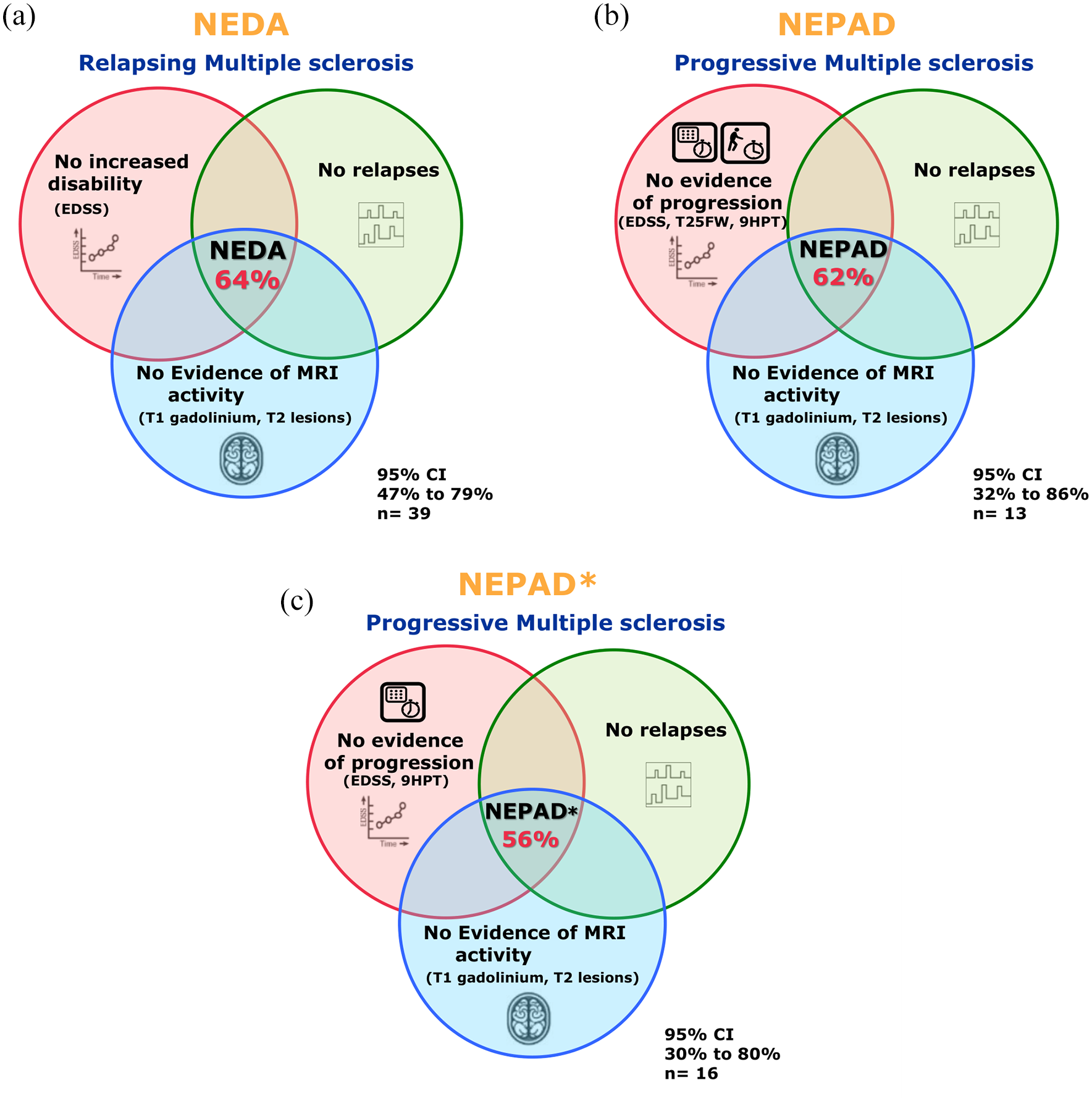
Composite measures indicating effectiveness of Litak® in patients with relapsing and progressive multiple sclerosis. (a) Venn diagrams representing the proportion of patients achieving No Evidence of Disease Activity (NEDA), (b) No Evidence of Progression or Active Disease (NEPAD), and (c) NEPAD excluding the Timed-25-foot-Walking-Test (NEPAD*). CI, confidence interval.
In 27 of 100 patients with RMS, 9HPT measurements were obtained at baseline and follow-up (median 13 months; range: 12–14). Dominant hand 9HPT times at baseline and follow-up were 18.0 and 18.9 s, respectively. Nondominant 9HPT times at baseline and follow-up were 20.9 and 20.2 s, respectively. Median T25FW times at baseline and follow-up (median 13 months; range: 12–14) were 4.9 and 4.1 s (n = 24).
In 31 out of 108 patients with PMS, 9HPT measurements were obtained at baseline and follow-up (median 13 months; range: 12–14). Dominant hand 9HPT times at baseline and follow-up were 25.0 and 26.3 s, respectively (n = 30). Nondominant 9HPT times at baseline and follow-up were 34.0 and 34.5 s, respectively (n = 31). Median T25FW times at baseline and follow-up (median 13 months; range: 12–16) were 7.6 and 6.8 s (n = 15).
NEDA, NEPAD, and NEPAD*
The proportion of patients with RMS having NEDA (total n = 39) at 18 months (median; range: 16–22) was 64% (95% CI: 47%, 79%; Figure 5). The proportion of patients with PMS (pwPMS) achieving NEPAD (total n = 13) at 19 months (median; range: 15–25) was 62% (95% CI: 32%, 86%). The proportion of pwPMS achieving NEPAD∗ (total n = 16) was 56% (95% CI: 30%, 80%).
Neurofilament light chain level in the cerebro-spinal fluid
In 23 out of 208 pwMS (n = 8 pwRMS and 15 pwPMS), CSF-NfL results were obtained before and after treatment. In pwRMS, CSF was obtained at least 3 months after their last documented relapse in all but one case. 27 Baseline lumbar punctures were performed 48 days (median; IQR: 12, 178) prior to starting treatment. Follow-up CSF was collected 311 days (median; IQR: 311, 357) after treatment initiation. Thus, 19 of 23 patients had one course, and 3 of 23 two courses of Litak® completed at the time follow-up CSF was collected. One patient had their follow-up LP between weeks 1 and 5 of their second treatment course.
CSF-NfL levels in the current cohort were 652 pg/mL (mean; IQR: 458–1063) and 334 pg/mL (mean; IQR: 186–505) at baseline and follow-up, respectively (p < 0.01; Wilcoxon signed rank test). Of the 13 out of 23 pwMS whose CSF-NfL was above ULN at baseline, 10 had normal levels at follow-up (Figure 6). In all three patients where CSF-NfL did not fall below their age-adjusted ULN (see below), NfL nevertheless dropped significantly in two of three from extremely high baseline levels (by 73% and 80%), after a single course of Litak®. 12

Cerebro-spinal fluid neurofilament light chain (CSF-NfL) levels in 23 patients with MS before and after treatment with Litak®.
Discussion
Whilst the availability of highly effective DMT has significantly improved for patients with RMS, 28 access to treatment for many pwMS remains limited, including patients with PMS and an EDSS of ⩾6.5. 14 Using Litak® off-label enabled treatment decisions based on the biology of MS as an immune-mediated disease throughout its course, 29 rather than cost-effectiveness criteria forming the basis of DMT eligibility. In particular, providing Litak® on a compassionate basis enabled the collection of preliminary data on the feasibility, safety, and potential effectiveness of a potent immunotherapy in people with advanced PMS. To identify treatments for this cohort of patients is among the top priorities of MS charities Worldwide.30,31
Following on from our early experience with Litak®, 9 we have now gathered data in a relatively large cohort of MS patients across the disease spectrum, heterogeneous in terms of age, disease course and duration, disability level, and prior interventions. The majority (n = 116) moved from a different immunotherapy to Litak®. In 77 of 116 patients (66%), transition to treatment with Litak® commenced within less than 6 months (Table 2), a time lapse short enough to be considered a ‘switch’ period between immunotherapies.
Two of our patients died in the context of receiving Litak®. In both cases, a causal relationship remains uncertain. While the treatment may have contributed to the risk of contracting H1N1 influenza in patient 1, TLC was within normal range 7 weeks before the patient’s rapid decline and death. Unfortunately, we were unable to obtain more detailed information about patient 2 who reportedly died with encephalitis of unknown cause, 44 weeks after completing his second course of Litak®.
Skin-related AEs, including shingles, were seen in 6%, including three previously reported cases of SASR. 21 A recent report on 239 pwMS treated with CladT detailed further skin reactions, and an overall incidence of 32%, 32 such that we conclude Litak® does not harbour a higher risk of skin pathologies than the licenced oral compound or indeed other highly effective immunotherapies used in MS. 33 Other AEs were rare. Of note, headaches were uncommon. While the phase III CLARITY trial of CladT in pwRMS reported headache in 21–24% (placebo: 17%), 23 head pains were detected in less than 3% in our cohort.
The large majority of pwMS underwent two courses of Litak®. Adjustment of the dose administered not only to body weight but also to individual TLC had the expected effect of reducing the number of patients with severe (grade 3) lymphopenia to 5%. Our safety-driven approach led to dose-adjustments based on low TLC in 25% during course 1 and 65% during course 2, with variable dose reductions.
We and others have previously suggested that depletion of memory B cells is crucial for effective disease control in pwMS.3,34 Evidence suggests that a single course of Litak® leads to significant and long-term depletion of B-cell subsets, particularly memory B cells, for at least 12 months. 4 Very similar results have recently been reported in patients with RMS treated with CladT 35 in the context of the MAGNIFY-MS study (NCT04783935). Of note, ongoing depletion of memory B cells occurred while other, particularly naive B-cell subsets, either normalised or exceeded their baseline level.4,35
We therefore expect that memory B cells remain depleted for at least 12 months after dosing with either parenteral 4 or oral35,36 cladribine. Further follow-up studies, including T-cell subsets, are underway to confirm (or reject) this hypothesis. However, follow-up data from a cohort receiving only a single course of CladT suggest that even half of the licenced dose bears significant effectiveness. An observational cohort of 90 pwRMS with a baseline median EDSS of 5.25 receiving one course of CladT remained stable on the EDSS for 2 years in 80% of cases. 37
The proportion of patients with RMS achieving NEDA following treatment with Litak® presented here is similar to the rate reported with CladT. 38 Our results also map onto findings in a recent Real World Evidence (RWE) cohort of n = 65 pwMS treated with either i.v. or oral cladribine. 39 Although comparison of results is limited since the authors of that study did not distinguish between RMS and PMS in their analysis of NEDA, the total cladribine dose administered and, in those treated with the i.v. preparation, adjustment to TLC indicates a similar therapeutic approach to ours. 39
The NEPAD rate in our cohort of PMS was in line with earlier evidence suggesting Litak® as a promising immunotherapy not only in RMS but also PMS.12,20,39,40 In order to include pwMS unable to complete the T25FW, we introduced NEPAD∗ removing the walking assessment. Both NEPAD and NEPAD∗ data suggest a positive effect of Litak® in more than half of the patients with available baseline and follow-up data (Figure 5(b) and (c)). However, the number of patients with full follow-up datasets is small, and conclusions regarding efficacy in this cohort should therefore be made with caution. Moreover, sensitivity analysis revealed that patients with higher EDSS scores were more likely lost to follow-up (Supplementary Table 1) thereby introducing bias. Having said that, we did not detect significant differences in characteristics of pwRMS who had complete datasets to calculate NEDA versus those without (Supplementary Table 2).
Common reasons for limited follow-up data in RWE cohorts also applied to our dataset: (1) MRI was performed as part of routine care provision. While acquisition protocols were not standardised across referring sites, they all included sequences routinely used in the care of patients with MS. Efforts are currently being undertaken to homogenise MRI protocols for pwMS across the UK.41,42 (2) Time constraints precluding collection of a hands-on EDSS during routine NHS appointments. Although not used in the current cohort, we recently started using a web-based version of the EDSS, 43 which we expect will lead to a more complete dataset at the next cut-off. (3) Phased introduction of systematic 9HPT and T25FW data collection only since late 2016 16 limiting the number of data points available for NEPAD/NEPAD∗. While no firm conclusions can therefore be drawn, our data9,14 underpinned the case for a placebo-controlled, multicentre trial of CladT in PMS and an EDSS of 6.5-8.5, which is now underway. 44 Our off-label cohort will remain under long-term follow-up including clinical decision-making regarding further DMT following two courses of Litak® treatment.
Supplemental Material
sj-docx-1-tan-10.1177_17562864211057661 – Supplemental material for Subcutaneous cladribine to treat multiple sclerosis: experience in 208 patients
Supplemental material, sj-docx-1-tan-10.1177_17562864211057661 for Subcutaneous cladribine to treat multiple sclerosis: experience in 208 patients by Kimberley Allen-Philbey, Stefania De Trane, Zhifeng Mao, Cesar Álvarez-González, Joela Mathews, Amy MacDougall, Andrea Stennett, Xia Zhou, Ozlem Yildiz, Ashok Adams, Lucia Bianchi, Camilla Blain, Christine Chapman, Karen Chung, Cris S Constantinescu, Catherine Dalton, Rachel A Farrell, Leonora Fisniku, Helen Ford, Bruno Gran, Jeremy Hobart, Zhaleh Khaleeli, Miriam Mattoscio, Sue Pavitt, Owen Pearson, Luca Peruzzotti-Jametti, Antonio Scalfari, Basil Sharrack, Eli Silber, Emma C Tallantyre, Stewart Webb, Benjamin P Turner, Monica Marta, Sharmilee Gnanapavan, Gunnar Juliusson, Gavin Giovannoni, David Baker and Klaus Schmierer in Therapeutic Advances in Neurological Disorders
Supplemental Material
sj-docx-2-tan-10.1177_17562864211057661 – Supplemental material for Subcutaneous cladribine to treat multiple sclerosis: experience in 208 patients
Supplemental material, sj-docx-2-tan-10.1177_17562864211057661 for Subcutaneous cladribine to treat multiple sclerosis: experience in 208 patients by Kimberley Allen-Philbey, Stefania De Trane, Zhifeng Mao, Cesar Álvarez-González, Joela Mathews, Amy MacDougall, Andrea Stennett, Xia Zhou, Ozlem Yildiz, Ashok Adams, Lucia Bianchi, Camilla Blain, Christine Chapman, Karen Chung, Cris S Constantinescu, Catherine Dalton, Rachel A Farrell, Leonora Fisniku, Helen Ford, Bruno Gran, Jeremy Hobart, Zhaleh Khaleeli, Miriam Mattoscio, Sue Pavitt, Owen Pearson, Luca Peruzzotti-Jametti, Antonio Scalfari, Basil Sharrack, Eli Silber, Emma C Tallantyre, Stewart Webb, Benjamin P Turner, Monica Marta, Sharmilee Gnanapavan, Gunnar Juliusson, Gavin Giovannoni, David Baker and Klaus Schmierer in Therapeutic Advances in Neurological Disorders
Footnotes
Acknowledgements
The authors are grateful for support by the clinical BartsMS team based at The Royal London Hospital including Aine Redfern-Walsh, Charlotte Sellers, Emma Ridgway, Freya Edwards, Grace Anjorin, Tatjana Sayali, and Thamanna Begum. For critical input and support of this service, and/or referring patients for consideration of treatment with Litak®, we would like to thank Jeremy Chataway, Siobhan Leary, and Wallace Brownlee (The National Hospital for Neurology and Neurosurgery Queen Square, University College London Hospitals NHS Foundation Trust); Heather Wilson and Gerard Davies (Royal Free London NHS Foundation Trust); Peter Brex (King’s College Hospital Foundation Trust); Victoria Williams (Guy’s and St. Thomas’ NHS Foundation Trust); Josephine Swanton (London North West University Healthcare NHS Trust); David Lieberman (Barts Health NHS Trust); Jeban Ganesalingam, Waqar Rashid and Romi Saha (Brighton and Sussex University Hospitals NHS Trust); Laura Petzold (Maidstone and Tunbridge Wells NHS Trust); David Rog (Salford Royal NHS Foundation Trusts); Nikos Evangelou (Nottingham University Hospitals NHS Trust), Gordon Mazibrada (University Hospitals Birmingham NHS Foundation Trust); Carolyn Young (The Walton Centre NHS Foundation Trust); Abhijit Chaudhuri (Barking, Havering and Redbridge University Hospitals NHS Trust) and Emma Gray (MS Society UK).
Author contributions
KS, DB and GG conceptualized and designed the study. GJ advised on the dosing schedule used. KAP, SDT and AS gathered the data. KAP and AM undertook the analysis. KAP and SDT drafted the the manuscript. All authors revised the manuscript for important intellectual content and approved the final version.
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: K.A.P., S.D.T., Z.M., A.M., A.S., X.Z., O.Y., A.A., L.B., C.C., C.D., S.P., L.P.J., B.S. and S.W. have no conflicts of interest to declare. C.A.G. is a founder of NeuroCreare Ltd. J.M. has received honoraria and meeting support from Arvelle, Biogen, Novartis, Merck Serono, Roche and Sanofi Genzyme. C.B. has received travel costs and honoraria from Novartis, Genzyme, Teva and Biogen. K.C. has received honoraria and travel grants from Biogen, Sanofi-Genzyme and Roche. C.S.C. has received support for research, attendance of conferences, and consultancy from Biogen, GW Pharmaceuticals, Novartis, Teva, Merck, Morphosys, Roche, Sanofi Pasteur MSD and Sanofi Genzyme. R.A.F. has received honoraria and consultancy fees from Merck, TEVA, Novartis, Genzyme, GW Pharma, Allergan, Merz, Ipsen, and Biogen. R.A.F.’s current research activity is supported by the NIHR Biomedical Research Centre UCLH. L.F. has received consultancy fees from Biogen, Novartis, Roche and Genzyme. L.F. has received support for educational events from Biogen, Genzyme, Merck, Novartis, Teva, and the Neurology Academy. H.F. has received support from the Health Technology Assessment Programme (NIHR) and the UK MS Society. In the past 3 years, H.F. has been a local principal investigator for trials in MS funded by Novartis, Roche, and Biogen Idec and has taken part in advisory boards and consultancy for Biogen Idec, Merck, Novartis and Roche. B.G. has received personal compensation for consultancy from Merck, Roche, Biogen, Teva UK, and GW Pharma. B.G. has received unrestricted research grants from Biogen Idec, Merck, Bayer Healthcare, Teva UK, Novartis, and Genzyme. B.G. has received support for the attendance of clinical and research conferences from Biogen, Merck, Bayer Healthcare, Teva UK, Novartis, Genzyme, and CelGene. J.H. has received consultancy fees, meeting support, or grants to support clinical services or research from: Biogen Idec, Sanofi Genzyme, Janssen Cilag, Merck, Neurodiem, Novartis, Roche, Celegene, Oxford pharmagenesis. Z.K. has received honoraria and travel costs from Roche, Biogen and Novartis. M.M. has received travel support and speaker honoraria from Biogen Idec, Genzyme, Merck-Sereno, Novartis, Roche and Teva and consultation for Celgene, Merck-Serono, Novartis and Roche. O.P. has received speaking fees and travel expenses from, and/or served on advisory boards for, Biogen, Bayer, Celegene, Janssen, Merck Novartis, Roche, Sanofi and Teva. A.S. has received honoraria, travel grants and been a member of advisory boards for Biogen, Novartis, Teva, Celgene, Sanofi, and Merck. E.S. has received consulting fees and/ or support to attend academic meetings from Merck. E.T. has received honorarium for consulting work from Novartis, Merck, Biogen, and Roche. E.T. has received travel grants to attend or speak at educational meetings from Biogen, Merck, Roche, Takeda, and Novartis. B.P.T. has received honoraria, travel grants, and been a member of advisory boards for Biogen, Merck Serono, Novartis, Sanofi Genzyme, and Roche. M.M. has received honoraria and travel costs from Genzyme, AbbVie, Roche, and Novartis. S.G. has received honoraria from Biogen Idec, Sanofi Genzyme, Janssen Cilag, Merck, Neurodiem, Novartis, Roche, and Teva and grant support from ECTRIMS, Genzyme, Merck, National MS Society, Takeda, and UK MS Society. G.J. has received speaker honoraria from and is a member of advisory boards of AbbVie, Astellas, Celgene, and Novartis. G.G. has received honoraria and meeting support from AbbVie Biotherapeutics, Biogen, Canbex, Ironwood, Novartis, Merck, Merck Serono, Roche, Sanofi Genzyme, Synthon, Teva, and Vertex. He also serves as chief editor for Multiple Sclerosis and Related Disorders. D.B. has received compensation from InMuneBio, Lundbeck, Merck, Novartis, Rock, and Teva. K.S. has received research support from Biogen, Merck KGaA, and Novartis, speaking honoraria from, and/or served in an advisory role for, Amgen, Biogen, EMD Serono, Merck KGaA, Novartis, Roche, Sanofi-Genzyme, and Teva; and remuneration for teaching activities from AcadeMe, Medscape and the Neurology Academy.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported, in part, by Multiple Sclerosis of Great Britain & Northern Ireland grant 69/2017. A donation by the Morris-Saady Charitable Trust is gratefully acknowledged. Z.M. has been supported by a 2016/17 ECTRIMS Clinical Training fellowship. R.A.F. has been supported in part by the National Institute for Health Research, University College London Hospitals, Biomedical Research Centre, London, UK.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.