
Abstract
Background:
Cerebral cavernous malformation (CCM), especially the familial form, is a relatively rare congenital and occult vascular disease of the central nervous system. The familial form of CCM has been linked to three different genes: KRIT1/CCM1, MGC4607/CCM2, and PDCD10/CCM3; however, the genetic basis of CCM is not well understood. The PDCD10/CCM3 is the most recent gene to be identified that results in worse clinical symptoms. Early diagnosis and treatment is important for patient prognosis.
Case report:
The proband is a 38-year-old male who has been suffering from weakness in the limbs for 7 months. Investigation of his family history revealed that his mother also suffered from limbs paralysis and had been bedridden for a long time. His older brother suffered from headache for years, whereas his younger brother was asymptomatic. Brain computed tomography analysis of all family members showed multiple high-density shadows. Subsequently, magnetic resonance imaging analysis identified more prominent and similar multiple intracranial lesions in all family members. The lesions were hypo-intense, or showed mixed signs on T1-weighted imaging, and were significantly more intense on T2-weighted imaging. To understand the genetic basis of the disease in the family, DNA sequencing analysis was performed. A novel deletion mutation in the PDCD10/CCM3 gene was identified in the proband and his relatives. The deletion resulted in a frameshift mutation and premature termination of translation of the protein, and potentially caused the disease in this family.
Conclusions:
Our study identified a novel PDCD10/CCM3 heterozygous deletion (c.165delT) associated with CCM. This finding expands the CCM gene mutation profile, which will be beneficial for genetic counseling and clinical therapy.
Introduction
Cerebral cavernous malformation (CCM) is classically defined as low-pressure, hamartomatous, and berry-like vascular lesions in the central nervous system. The prevalence of all types of CCM is predicted to be 0.1–0.5% in the general population.1,2 CCM occurs either as sporadic or familial; inheritance of the familial form is autosomal dominant, with variable expression and incomplete penetrance. 3 It is clinically silent, or associated with headache, hemorrhagic strokes, seizures, or focal neurological deficits. In addition to the symptoms, brain computed tomography (CT) and magnetic resonance imaging (MRI) are commonly used for examining CCM, with brain MRI being more sensitive in revealing lesions, especially on susceptibility weighted imaging (SWI). Images of CCM do not appear the same in all individuals due to the repeated episodes of bleeding in brain lesions. A typical image can be seen on T2-weighted MRI of the brain, which is characterized by a mulberry- or popcorn-like appearance in the center, surrounded by a dark rim. 4 CCM is caused genetically by loss-of-function mutations in one of the following three genes: KRIT1 (Krev1 Interaction Trapped 1)/CCM1, MGC4607 (malcavermin)/CCM2, and PDCD10 (Programmed Cell Death 10)/CCM3. To date, more than 100, 30, and 20 distinct mutations have been identified in CCM1, CCM2, and CCM3, respectively. 1 In our study, a novel mutation in the CCM3 gene was identified in a Chinese family. This finding widens our understanding of the mutation pools in genes associated with CCM, which will be useful for studying CCM in the future.
Case report
A 38-year-old male presented to our outpatient department with a 7-month history of left limbs weakness. He had no history of smoking, hypertension, diabetes, or any other risk factors for cerebrovascular diseases. Physical examination upon admission revealed paralysis of the left limbs and slight paralysis of the face. Babinski and Chaddock signs on the left side were positive, whereas other nervous system examination results were negative. Brain CT showed multiple high-density shadows, which indicated a tendency for calcification (Figure 1). For further diagnosis, MRI was performed, and more lesions were found with MRI compared with CT. The brain lesions were partly hypo-intense on T1-weighted imaging, whereas they were significantly more intense on T2-weighted imaging. The other lesions were hyper-intense in the center and surrounded by a hypo-intense rim on T1/T2-weighted imaging. SWI displayed more hypo-intense lesions, indicating hemorrhage (Figure 2). Investigation of the family history showed that his relatives also suffered from physical disorders, albeit to different degrees. His mother suffered from progressive limb paralysis, and had been bedridden for a long time. Her brain MRI showed leukoaraiosis and multiple hypo-intense lesions on T2/T1 and diffusion weighted imaging (DWI), indicating hemorrhage. His older brother had suffered from headache for several years without any symptoms of limb paralysis. His younger brother was asymptomatic. Although his brothers had different symptoms, we noted similar findings on the brain CT and MRI (Figure 3) scans. All observations indicated that the patient probably had a genetic disease.

Brain CT of the proband shows small high-density shadows in the cerebral peduncle (a), cortical/juxta-cortical region and corona radiata (b, c).

Brain MRI of the proband. The lesion of the cerebral peduncle is hyper-intense in the center and surrounded by a hypo-intense rim on T2-weighted imaging (a), and on T1-weighted imaging (b). The lesions of the cerebral cortex show mixed imaging on T2 and T1-weighted imaging (c, d). Brain lesions are more prominent on SWI, which shows multiple hypo-intense lesions (e, f).
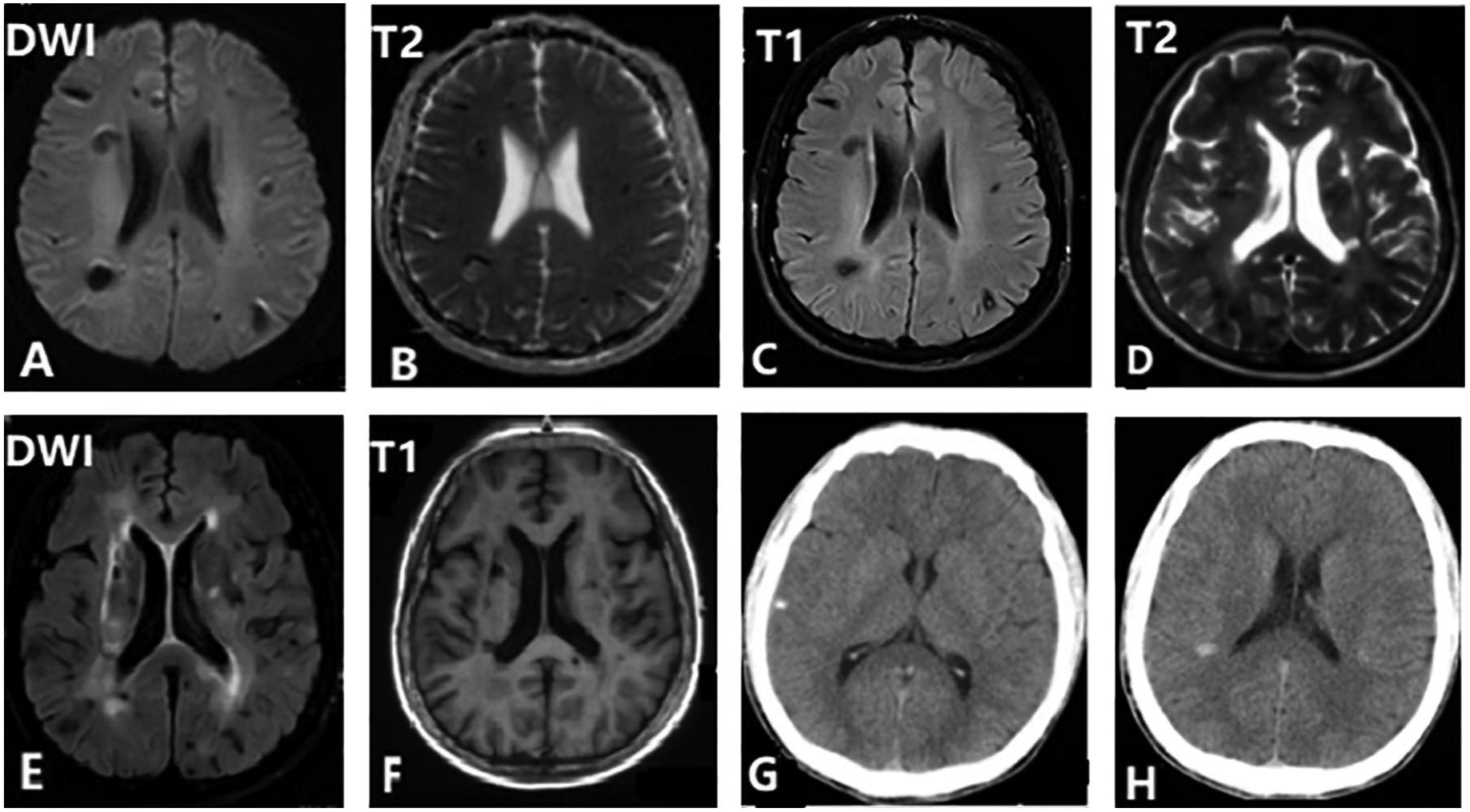
Brain MRI scans of the proband’s older brother (a–c) and his mother (d–f) show multiple lesions in the cerebral cortex and centrum semiovale. These lesions are hypo-intense or mixed on T2/T1-weighted imaging, and more prominent on DWI. Brain CT of the proband’s younger brother shows multiple high-density shadows (g, h).
Molecular genetics studies
To examine the genetic cause of the disease, after obtaining written informed consent from the family members, DNA sequencing was performed. Genomic DNAs were extracted from the peripheral venous blood of all the family members using a blood DNA extraction Kit (CWE9600). DNA was prepared in the laboratory using a KAPA Library Preparation Kit; sequencing was then performed using Illumina NovaSeq. Sequence analysis of the CCM1, CCM2, and CCM3 genes identified a novel heterozygous deletion in the CCM3 gene (c.165delT, p.N55fs) (Figure 4). The 165th nucleotide in the coding region of the CCM3 gene had a heterozygous deletion, which resulted in a frameshift mutation leading to prematurely terminated translation of the protein. The same mutation in the CCM3 gene was found in the proband’s brothers and mother, indicating that the mutation is associated with the disease in the family, and has not been reported before.

DNA sequencing analysis of the proband (a) showing the heterozygous deletion in the PDCD10/CCM3 gene: c.165delT, p.N55fs (red circle). The same mutation was found in his brothers and mother (b–d).
Treatment
So far, no medical therapy has been proven to be useful for CCM, especially for small multiple lesions. Symptomatic treatment, such as controlling blood pressure and relieving headache, is the treatment of choice for the proband and his relatives at present.
Discussion
CCM is a relatively rare and occult vascular disease of the central nervous system. The prevalence of all types of CCM is approximately 0.1–0.5%.1,2 The familial form of CCM is associated with three genes: KRIT1/CCM1, MGC4607/CCM2, and PDCD10/CCM3, of which CCM3 is the most recently discovered and has more severe disease manifestations compared with the other two.5,6 To date, there are ~20 CCM3 mutations that can lead to CCM. According to previous reports, most of the indels occur in the N-terminal region of CCM3 gene 7 ; however, the etiology, pathogenesis, and treatment of CCM are still not well understood. In our case, we found a novel CCM3 mutation associated with CCM that has not been reported before, which is potentially due to highly conservation of this site. The fact that all family members of the proband have different clinical manifestations, and have been suffering from the disease since an early age, is similar to a previous study reported by Kevin Whitehead and his team, wherein they state that patients with a CCM3 mutation have a greater chance of spontaneous hemorrhage, which presents at a younger age. 8 All clinical manifestations of the proband and his family members indicate that the CCM3 mutation may lead to severe clinical symptoms and poor prognosis.
So far, several gene mutations have been found to be associated with CCM in different populations. Notably, most of these gene mutations are heterozygous and are either frameshift, nonsense, or splice site mutations that result in different forms of the protein and eventually lead to CCM. 9 In our study, DNA sequence analysis of the proband and his relatives revealed the presence of a deletion of T at the 165th position in the coding region of the CCM3 gene. This heterozygous deletion changes the nucleotide sequence and leads to a frameshift in the gene. Consequently, protein translation is terminated prematurely. The clinical evidence and family history investigation reveal that the gene mutation is the cause of the disease in this case. However, the exact pathogenic mechanism is still unclear. Recently, studies have shown that the CCM3 gene may be associated with cell apoptosis, and is involved in the CCM1/2 pathway that is associated with the disease. 10 Products of the CCM3 gene play major roles in angiogenesis and endothelial cell junctions in neural tissue. Loss-of-function mutations in the CCM3 gene lead to extensive vascular abnormalities and increased permeability. 11 Another study showed that oxidative stress and inflammation may have an important impact on the progress of CCM. 12 Due to the limited numbers of CCM carriers, more studies are required to investigate the problem.
With the development of imaging technology, diagnosis of CCM has become easier than previously. CCM usually cannot be noticed through angiography. MRI has near-perfect sensitivity and excellent specificity, and, therefore, is the best available option for the detection and characterization of CCM. 13 Generally, the appearance of CCM lesions on MRI scans differs from case to case. A typical image is presented as a mixed signal on both T1- and T2-weighted imaging. On T2-weighted imaging, the lesions have a hypo-intense rim, resulting in a ‘popcorn-like’ appearance; however, physiologic senile iron deposition will also cause hypo-intense lesion on normal brain.11,14 In our study, the proband and his relatives had similar MRI manifestations; the brain lesions were partly hypo-intense on T1-weighted imaging, whereas they were significantly more intense on T2-weighted imaging. Other lesions were hyper-intense in the center, and surrounded by a hypo-intense rim on T1/T2-weighted imaging. The reason we did not observe typical images in our case may due to whether or not the lesions are hemorrhagic, and the time of the hemorrhage. It is possible that the differences in manifestation are associated with the underlying gene mutation. The proband also underwent complete SWI, which showed more lesions, and these lesions were more prominent than those seen on conventional MRI. This confirms the idea that brain SWI can accurately recognize deoxyhemoglobin and hemosiderin, and, therefore, is advantageous for the detection of CCM lesions. 13 The diameter of the CCM lesions ranges from a few millimeters to several centimeters, and new lesions appear at a rate of 0.2–0.4 lesions per patient-year. The accuracy of brain MRI makes it a suitable method for the evaluation of brain lesion development. 15 With the development of imaging technologies, diagnosis of CCM will be quicker and more accurate.
The current treatment for CCMs is surgical resection, and no other medical therapies have been approved so far. Laboratory and preclinical studies have shown that statins are a potential therapy for CCM 8 ; however, more studies need to be done in the future. Based on studies of the CCM mechanism, some drugs have been tested in animals. In our case, the proband was suggested to take statins and to control his blood pressure. Follow up on further curative effects will be required.
Conclusion
CCM is a rare vascular disease of the central nervous system. In this study, we identified a novel heterozygous deletion in the CCM3 gene. This finding expands the CCM gene mutation profile, and will be beneficial for genetic counseling. However, further laboratory studies and clinical assessments are necessary to identify the pathogenesis of CCM and potential targets for therapy.
Footnotes
Acknowledgements
The authors thank Editage for proofreading the manuscript.
Ethics statement
Written informed consent for publication of the case report was obtained from the patient and his relatives.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and publication of this article: This work was supported by the National Natural Science Foundation of China (No, 81601088; No, 81974191).
Conflict of interest statement
The authors declare that there is no conflict of interest.