
Abstract
This case report examines a newborn with bilateral postaxial polydactyly type B, delivered by a 42-year-old mother with a history of third-degree consanguinity. The mother, having had no prior live births and one abortion, presented at 39 weeks gestation. The absence of prenatal care is noted, with its potential impact on prenatal diagnosis not assessed. The newborn, a healthy girl, weighed 3400 g with an Apgar score of 9/10. Radiographic and physical examination revealed vestigial sixth digits with rudimentary phalanges, influencing the surgical approach. This report underscores the importance of genetic counseling in cases of consanguinity and illustrates the multidisciplinary strategy necessary for managing polydactyly, from surgical considerations to genetic evaluation.
Keywords
Introduction
Polydactyly, derived from the Greek words “poly” (many) and “dactylos” (digits), refers to a congenital condition characterized by the presence of extra fingers or toes. This anomaly is one of the most common congenital deformities of the hand and foot, observed in approximately one out of every 1000 live births. 1
Polydactyly can manifest in two primary forms: nonsyndromic, where the extra digits appear in isolation, and syndromic, where they are part of a broader constellation of congenital defects. 2 The condition shows a notable predilection for certain limbs; it is more common in the right hand compared to the left, and more frequently, affects the upper limbs than the lower, and occurs more often in the left foot than in the right.1,3,4
The clinical presentation of polydactyly can vary significantly, ranging from fully formed and functional digits to rudimentary, nonfunctional appendages. This variability in clinical manifestations presents specific challenges and considerations in treatment planning. This case report discusses the management of a newborn diagnosed with polydactyly, along with the challenges and considerations associated with treatment.
Case report
This case report describes a 42-year-old woman, G2P0A1, indicating her second pregnancy with no prior live births and one abortion. She has a history of third-degree consanguinity, being a product of first cousins, which may contribute to genetic predispositions. Both the patient and her partner are nonsmokers. The patient presented at 39 weeks of gestation experiencing labor pains.
Upon examination, the outcome was the delivery of a healthy baby girl, weighing 3400 g with an Apgar score of 9/10, which reflects the newborn’s excellent physical condition shortly after birth.
A notable finding in the newborn was bilateral postaxial polydactyly type B (PAPB). This form of polydactyly is characterized by the presence of extra digits that lack substantial bone structure, typically presenting as soft tissue extensions. Physical examination revealed the presence of nails on the vestigial digits, suggesting some form of underlying cartilaginous support despite the absence of bones. X-ray imaging supported the absence of bony structures in these digits but could not definitively rule out the presence of cartilaginous elements.
The case report further discusses the clinical implications of such a finding. Given the absence of familial history of polydactyly, this case suggests a potential de novo mutation or an unrecognized genetic trait within the family. This emphasizes the importance of genetic counseling for families with similar histories of consanguinity.
The surgical team recommended a specific approach for the amputation of the extra digits. Due to the predominantly soft tissue composition, a meticulous surgical technique using fine dissection methods was employed to ensure minimal scarring and to preserve maximum functionality and sensation in the affected areas. This strategy reduces the risk of potential complications and aligns with the best practices for such congenital conditions (Figure 1).
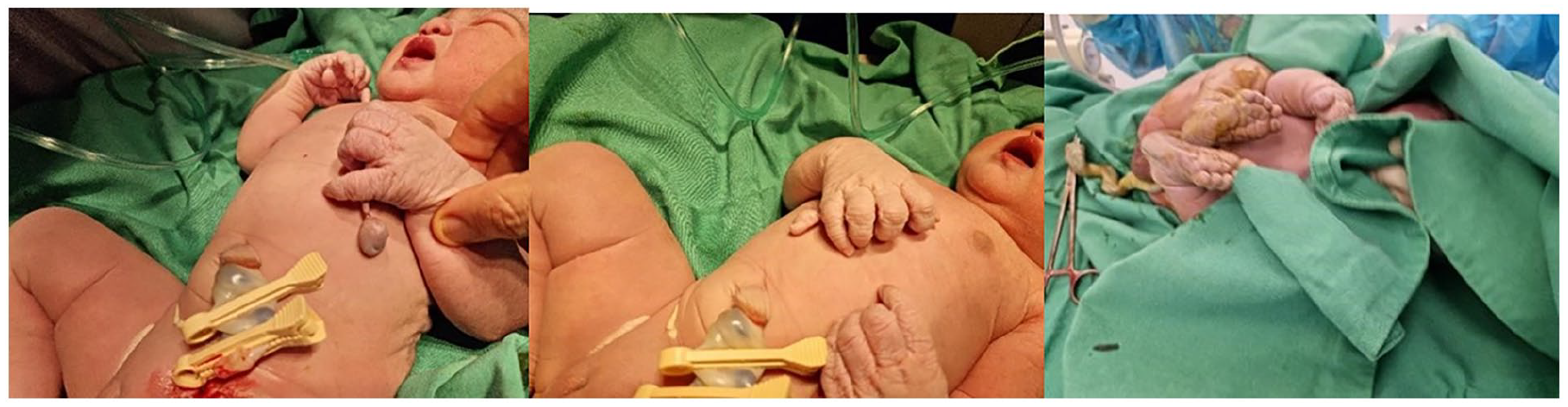
Photograph of the newborn baby girl upon delivery presenting bilateral upper limb postaxial polydactyly type B with vestigial sixth digits along with normal lower limb digits.
This detailed exploration of bilateral postaxial polydactyly highlights the specific challenges and considerations in treatment planning, underscoring the need for a multidisciplinary approach to effectively manage and address both the surgical and genetic aspects of polydactyly.
Discussion
Polydactyly, the presence of extra digits, exhibits varying incidence rates across different ethnic and demographic groups, which suggest a strong genetic component influenced by ancestral background. According to a study by Finley et al., polydactyly is significantly more prevalent among African-American populations compared to Caucasian populations in the United States, with rates as high as 13.5 per 1000 in men and 11.1 per 1000 in women of African descent. 5 This contrasts sharply with rates in men and women of likely European descent, which are 2.3 per 1000 and 0.6 per 1000, respectively. Such statistics underscore the need for understanding genetic predispositions in different ethnic groups to better anticipate and manage this congenital anomaly.
Polydactyly is classified into several systems that help delineate treatment strategies and understand embryological origins. The Swanson classification, introduced in 1976, categorizes congenital hand deformities into seven types (Table 1), with Type III pertaining to duplications like polydactyly. 6 In parallel, the minimally access surgery (MAS) classification focuses more on the surgical and anatomical perspective, essential for operative planning. It classifies polydactyly based on the presence and type of tissue connections, with Type A indicating soft tissue attachments. 7
Classification of hand abnormalities by Swanson.

Source: Adapted from. 6
More recently, the Oberg, Manske, and Tonkin (OMT) classification was adopted by the International Federation of Societies for Surgery of the Hand, which categorizes hand anomalies into malformations, deformations, dysplasias, and syndromes.8,9 When it comes to the malformations category, two major subdivisions exist: (A) entire upper limb and (B) handplate. The latter usually involve early limb patterning and late limb patterning/differentiation, respectively. The OMT classification further subdivides these malformations based on the primary axis involved: (a) Proximodistal axis, (b) Radioulnar (anteroposterior [AP]) axis, (c) Dorsoventral axis and (d) Unspecified axis. In the context of our patients, ulnar polydactyly was noted which is a malformation of the handplate along the radioulnar (AP) axis according the OMT classification.10,8 Indeed, this scheme assists in the multidisciplinary approach to congenital abnormalities, promoting a more tailored therapeutic strategy based on the type of anatomical abnormality present. Table 2 further highlights the different classifications of hand abnormalities met by the patient in this case study.
Different classifications of hand abnormalities met by the patient in this case study.

PAPB: postaxial polydactyly type B.
Polydactyly may be caused by disruptions in the programmed cell death that is crucial during fetal limb development before the 8th week of gestation or by genetic mutations in the zone of polarizing activity, which is responsible for limb development.11,12 Particularly, mutations in the Sonic Hedgehog (SHH) gene, a critical component of the SHH signaling pathway for limb patterning, can lead to aberrant digit formation.13,14,15 This condition can manifest as either nonsyndromic, appearing without other congenital anomalies,16,17 or syndromic, occurring alongside other abnormalities and possibly linked with several syndromes such as Fanconi anemia, VACTERL, trisomy 13, and trisomy 21.18,2,3,19
Nonetheless, it is also important to note that Turing activation-diffusion patterning mechanism is also involved in digit development as it defines Sox9-positive digits and Bone Morphogenetic Proteins (BMP)-positive interdigital regions. Indeed, the current body of literature highlights how a Turing network implemented by BMPs, Sox9, and Wnt signaling pathways does contribute to driving digit specification. Particularly, when moderated by morphogen gradients, a self-organizing Turing network can recapitulate the expression patterns of Sox9, thus resulting in reproducible pattern formation of digits.20,21
On another level, it is also worth mentioning that the gradient established by Shh, along with its downstream transcription factor Gli3, does fine-tune this Turing network. In particular, Gli3 was proposed to negatively regulate Shh. Gli3 achieves this regulation by restricting the expression of Shh along with its influence on the posterior mesoderm. The current body of literature has noted that Shh and Gli3 are dispensable for formation of limb skeletal elements. The study of Litingtung et al., further investigates these genetic components in mice and demonstrates that Shh−/− Gli3−/− limbs are distally complete and polydactylous, but completely lack wild-type digit identities. 22
In addition to the Shh-GLI3 axis, the Apical Ectodermal Ridge (AER) and Hoxd10-13 genes also contribute to the regulation of digit number and identity. The AER produces Fibroblast Growth Factors that interact with Hoxd genes to modulate digit pattern, thus influencing the number and size of digits formed. 21
At the genetic level, postaxial polydactyly exhibits significant complexity and diversity. Research has identified multiple loci on chromosomes 7, 13, and 19 that are specifically linked to postaxial polydactyly. In total, at least 10 loci and 7 genes have been implicated in nonsyndromic forms of postaxial polydactyly1,23,24,25 (Figure 2). This genetic variability contributes to the wide range of phenotypic expressions seen in polydactyly, from fully functional digits to nonfunctional, rudimentary nubbins. These findings are reflective of the diverse genetic pathways that can lead to polydactyly. Accordingly, it remains important to understand this condition’s complexity while assessing the need for thorough genetic screening and counseling in affected families to better understand the potential inheritance patterns and associated risks.
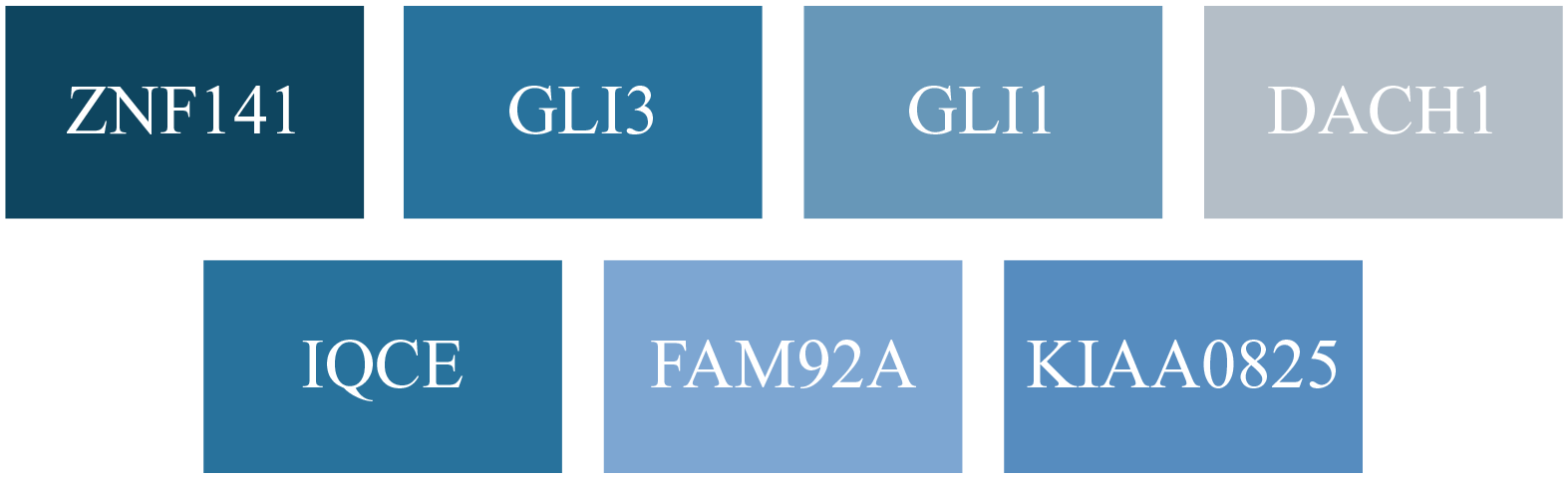
Genes involved in nonsyndromic postaxial polydactyly.
Postaxial polydactyly manifests differently depending on genetic and anatomical factors, which are influenced by ethnicity and specific mutations. Radial polydactyly, primarily seen among individuals of likely European descent, often occurs in a sporadic form on the hand anterior to the thumb. In contrast, ulnar polydactyly, as described by Temtamy and McKusick, is classified into two main types: Type A and Type B. Indeed, Type A involves well-formed digits with a bony connection to the hand and is subdivided into ten categories (A1-A10) (Table 3). In contrast, Type B refers to an incompletely formed, nonfunctional digit known as pedunculated postminimus, characterized by a narrow neurovascular pedicle without any bony connection.26,27,25
Forms of postaxial polydactyly along with their associated loci, mode of inheritance, and associated genes.

AD: Autosomal Dominant; AR: Autosomal Recessive; PAPB: postaxial polydactyly type B.
The gene Gli3, located on 7p14.1, has been identified as a key factor in postaxial polydactyly, mainly illustrating the genetic diversity and complexity of this condition.23,24,25 In particular, GLI3 mutations are associated with PAPB, the postaxial polydactyly recognized in the presented case.
Despite advancements in prenatal imaging, polydactyly often remains undiagnosed until birth, possibly leading to unexpected findings in the delivery room. It is possible, however, to detect fetal finger buds via transvaginal ultrasound as early as 9 weeks of gestation. If polydactyly is observed, further evaluation for congenital anomalies and associated syndromes is recommended, followed by a detailed biometric profile ultrasound between 17 and 34 weeks of gestational age if no additional anomalies are detected.2,30,31
Postnatal management requires detailed anatomical assessments using radiologic techniques, such as AP and lateral radiographs, to determine the exact structure of the extra digits and guide surgical decisions.2,32 In cases where the extra digits involve only skin connections, a nonsurgical approach like suture ligation may be used to stop the blood flow, causing the digit to naturally fall off.33,34 More complex cases, involving bony connections, ligaments, and tendons, typically require surgical intervention to prevent functional impairment and achieve cosmetic improvement.
For type A ulnar polydactyly, the management may involve suture removal under local anesthesia; however, this can lead to painful neuroma formation; hence, sharp excision might be employed to prevent such complications. For Type B, where the digit is less-developed, suture ligation is often sufficient33,34, while some practitioners also opt for primary surgical excision due to growing concern over short- and long-term complications 35 (Table 4).
Comparative table between type A and type B ulnar postaxial polydactyly.

Limitations
One of the limitations of this case report is the absence of long-term follow-up data. Due to logistical constraints and the ongoing healthcare crisis in the patient’s region, consistent postnatal follow-up could not be conducted as ideally required. This limitation restricts our ability to provide a comprehensive outlook on the prognosis and long-term management of polydactyly in this particular case.
Conclusion
Polydactyly can be transmitted through autosomal dominant or autosomal recessive (AR) inheritance, with AR inheritance posing a greater risk in children of consanguineous parents. It is critical to educate parents on the importance of prenatal follow-up and ultrasonography, not only to detect polydactyly but also to screen for associated congenital anomalies and syndromes that might accompany this condition. Early detection is essential for effective management. Upon diagnosis of polydactyly, an interdisciplinary team plays a crucial role in providing comprehensive support. This team helps families understand the implications of the condition, discusses relevant family history and consanguinity, and offers counseling about the possible outcomes for the affected child and the available treatment options. Additionally, in cases where there is a recurrence of polydactyly in families, genetic counseling becomes a vital component of care, helping families assess their risk and make informed decisions about future pregnancies.
Footnotes
Acknowledgements
We extend our gratitude to our esteemed colleagues for their unwavering support and invaluable insights that significantly contributed to this research.
Author contributions
K.G. and W.A. spearheaded this case report encompassing the conception and design, data acquisition, analysis, and interpretation. They played a pivotal role in drafting the manuscript and provided final approval for the version to be submitted. C.E.H., M.M., and G.Y. further enriched the manuscript by critically revising it for significant intellectual content and also extended their approval for the final version to be submitted. C.M. played a crucial role in advising on the content, ensuring the medical accuracy, and enhancing the overall presentation of the manuscript. His substantial contributions in shaping and refining the paper warrant recognition as an author.
Data availability
The data supporting the findings of this study are available from Rafic Hariri University Hospital.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Ethical approval to report this case was obtained from RAFIC HARIRI UNIVERSITY HOSPITAL BY DR. GEORGES YARED.
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.