
Abstract
This review is an overview of systemic conditions that can be associated with peripheral nervous system dysfunction. Children may present with neuropathic symptoms for which, unless considered, a causative systemic condition may not be recognized. Similarly, some systemic conditions may be complicated by comorbid peripheral neuropathies, surveillance for which is indicated. The systemic conditions addressed in this review are critical illness polyneuropathy, chronic renal failure, endocrine disorders such as insulin-dependent diabetes mellitus and multiple endocrine neoplasia type 2b, vitamin deficiency states, malignancies and reticuloses, sickle cell disease, neurofibromatosis, connective tissue disorders, bowel dysmotility and enteropathy, and sarcoidosis. In some disorders presymptomatic screening should be undertaken, while in others there is no benefit from early detection of neuropathy. In children with idiopathic peripheral neuropathies, systemic disorders such as celiac disease should be actively excluded. While management is predominantly focused on symptomatic care through pain control and rehabilitation, some neuropathies improve with effective control of the underlying etiology and in a small proportion a more targeted approach is possible. In conclusion, peripheral neuropathies can be associated with a diverse range of medical conditions and unless actively considered may not be recognized and inadequately managed.
Introduction
Peripheral nervous system (PNS) dysfunction can complicate various systemic diseases. Whilst this is more common in the adult age group, children are also at risk. Children are exposed to multiple types of systemic dysfunction, especially from inflammation, secondary infections, toxins, and so on, which potentially lead to primary and secondary impact on the PNS.
Systemic involvement may be the presenting feature and an underlying neuropathy may not be detected unless screened for by targeted examination and neurophysiological studies. People with conditions such as lymphoma, diabetes mellitus and uremia are at risk of PNS dysfunction, which can be clinically striking and symptomatic. Patient management focuses on the systemic disease, with the risk that comorbid complications are missed.
In other situations, PNS dysfunction may be the instigator leading to presentation for medical assessment. In this setting exploration for an underlying systemic disease should be included as part of the diagnostic assessment.
This review focuses on systemic conditions affecting children, which may be associated with PNS dysfunction, but excludes peripheral nerve disease which is reported with neurodegenerative disorders, metabolic diseases, and infections. The key findings are summarized in Table 1. Recommended and explorative therapeutic interventions are assessed for each condition.
Systemic conditions associated with peripheral nervous system dysfunction in children.

Search strategy and methodology
Articles were searched for on PubMed with no date limitation and targeting human studies. Case reports were included for some conditions as these were the only resources with documented information. Where this occurred, it was noted in the text. Search terms included ‘child’ and ‘neuropathy’ as standard, and were linked to each of the system disease headings, which were searched for under the main title as well as each of their subconditions. For example ‘vitamin deficiencies’ was followed by searches under each type of vitamin deficiency, similarly ‘endocrine disorders’ was followed by a search under ‘thyroid disease’, ‘hypothyroidism’, ‘hyperthyroidism’, ‘diabetes mellitus’, and so on. Only conditions associated with peripheral neuropathy in children were included. Information, where available, was drawn from the text relating to prevalence, clinical features, neurophysiology, and histological findings as well as management and outcome. Studies on peripheral neuropathies associated with neurodegenerative disorders, metabolic diseases, and active infections were excluded.
Critical illness polyneuropathy
Critical illness polyneuropathy (CIP) occurs as a result of systemic inflammation in patients with sepsis, severe respiratory illnesses, after transplantation of organs, and multiple systemic failure of organs, as well as those requiring extracorporeal life support.1,2 The pathophysiology of CIP is poorly understood, but most probably includes a combination of microvascular (vasodilation, increased permeability), metabolic (hyperglycemia, mitochondrial failure, hypoalbuminemia) and electrical alterations (inactivation of sodium channels and change in resting membrane potential). 1 CIP is part of a group of disorders including critical illness myopathy (CIM) and combination-critical illness polyneuromyopathy (CIPNM). 1 The condition is rare in children, reported to affect 0.02% of pediatric intensive care unit (PICU) admissions, but this figure may be an underestimation. 2 Clinical presentation of CIP is often first considered following failure to wean from artificial ventilation support. 97 In the PICU environment the associated generalized weakness, muscle atrophy, and absent or reduced deep tendon reflexes may be difficult to detect. The condition can present within the first week of illness, with ranges between 4 and 26 days reported.97,98 Children affected by severe burns have been reported to suffer a condition similar to CIP. 99 Differential diagnoses of CIP, include spinal cord pathology, neuromuscular blockade, autoimmune myasthenia gravis, corticosteroid- or relaxant-induced myopathy, acute necrotizing myopathy, low blood phosphate levels, toxic and vitamin deficiencies (e.g. thiamine-deficiency), asthma-amyotrophy syndrome, and acute inflammatory demyelinating polyradiculoneuritis. Useful screening investigations include assessing magnesium and phosphate levels, serum creatine kinase levels, which are normal in most cases of CIP, neurophysiological studies, and relevant imaging modalities. Extensive muscle denervation and axonal degeneration is found on peripheral nerve studies and biopsy samples.1,3
Children who already have life-threatening conditions and are affected with CIP/CIM can suffer significant additional morbidity. 97 Supportive care is the mainstay of treatment; with prompt correction of impaired osmolar states, glycemic levels and electrolyte imbalance, in addition to early rehabilitation.4–6 Spontaneous recovery occurs but may be delayed. 97 The updated 2014 Cochrane review found moderate quality evidence that use of insulin therapy could reduce symptoms of CIP/CIM, and high-quality evidence to support that it was associated with reduced duration of artificial ventilation, duration on ICU and mortality at 180 days, but identified potential risks associated with hypoglycemia. 6 Corticosteroid therapy is ineffective in CIP/CIM. 6 There is moderate evidence that early rehabilitation of CIP/CIM may facilitate weaning from ventilatory support. 6 No beneficial effect was evident from electrical muscle stimulation, based on very low-quality evidence. 6
Chronic renal failure
Nerve conduction studies in children with long-term dialysis-dependent renal failure are usually either normal or mildly neuropathic. 7 Two-thirds of uremic children with abnormal common peroneal nerve conduction studies have no symptoms of peripheral neuropathy.8–10 Evaluation of the ‘H’ reflex is a useful marker in a child lacking clinical signs of uremic polyneuropathy. 11 As such this neurophysiological study is important to include in assessments as the complication is found in almost 60% of children with chronic renal failure. 11 An axonal degenerative neuropathy is usually found, although any type of neuropathy (purely axonal, mixed or predominantly demyelinating) may occur. 12
Symptomatic children may experience sensory abnormalities in the lower limbs; these can, over time, evolve into a sensorimotor polyneuropathy with flaccid paraplegia or quadriplegia. A purely motor type of uremic neuropathy is also reported. 100 Symptoms may be improved though longer periods and frequency of dialysis. Even in patients with established uremic neuropathy, which may be due to chronic hyperpolarization secondary to altered potassium levels, renal transplantation can lead to resolution of the complication. 13
Endocrine disorders
Polyneuropathy may also occur in patients with hypothyroidism 101 and hypoglycemia, 102 as well as with diabetes and multiple endocrine neoplasia (MEN).
Diabetes mellitus
Studies report a wide range, namely between 10% and 68%, of children with diabetes who are affected with neuropathy.14,15 One study of 146 children with diabetes found that whilst 27.4% had features of peripheral neuropathy, in 62.5% this was subclinical. 19 Diabetic neuropathy may manifest as a polyneuropathy, focal neuropathy, or autonomic neuropathy. 14 Autonomic neuropathy leads to the most serious clinical impact from lack of awareness of hypoglycemia and cardiovascular dysfunction, and is the most common neuropathy to occur. Rare cases of mononeuritis multiplex are also reported. 103 Whilst some affected patients believe themselves to be asymptomatic, their clinical assessment may identify sensory loss. Impaired vibrotactile sense is a useful clinical marker of neuropathy in children with diabetes. 104 Numbness was detected in 30.8% of subjects in one study, with large myelinated nerve fiber dysfunction found in 7–10%; pain or temperature sensation impairment was found in only 1.4%. 19 A length-dependent polyneuropathy in children may lead to distal sensory loss (numbness) and paresthesia with burning, aching, or pinprick dysesthesia. In addition, affected children may have motor dysfunction related to distal weakness and poor coordination as well as distal anhidrosis, post-eating bloating, constipation, diarrhea, and impaired awareness of hypoglycemia. Deep tendon reflexes may be absent and foot ulcers can occur. Mildly impaired autonomic nervous system function was reported in 30–50% of children with diabetes who were tested soon after diagnosis.105,106
Nerve conduction studies most often detect abnormal measurements from the peroneal and sural nerves.19,107,108 A large prospective study of nerve conduction and autonomic nervous system function in diabetic children detected slowing of sensory nerve conduction and impaired autonomic function in 25% of children at the time of diagnosis. 17 Of 50 juvenile diabetics with a mean age of 13 years, 10% had decreased median nerve motor conduction velocities, 32% had slowed conduction in the posterior tibial nerves, and 44% slowing of sural nerve conduction. 18 Diabetic neuropathy appears to be more marked in those with poorly controlled diabetes, but is also reported in young children with a short illness duration and good glycemic control. 16 Based on this, duration of illness cannot be stated to be a key disease-inducing factor. The nerve dysfunction can occur acutely and rapidly in the early stages from diabetes onset, although following this the neuropathy progresses more slowly and may even plateau. 19 The best preventative strategy for diabetic neuropathy remains good blood glucose control.19,20 Serial nerve conduction studies are recommended to identify onset of this high-impact disease complication. 109 Most interventional studies of the management of diabetic neuropathy are experimental, or in adults with type 2 diabetes. 110 The potential benefits of aldose reductase inhibitors, which act on enzymes in the polyol pathway involved in the metabolism of blood glucose, and benfotiamine, a fat-soluble analog of thiamine that in animal models inhibits vascular damage in diabetes, are of potential interest.111,112 C-peptide, the 31 amino acid component of proinsulin, may reverse the structural and functional changes due to diabetes in rats and humans. 113 It improved sensory function in patients with type 1 diabetes and mild neuropathy. 113 Angiotensin-converting enzyme inhibitors may also have a role. 110 For symptomatic relief of painful neuropathy most guidelines recommend use of tricyclic agents, serotonin–norepinephrine reuptake inhibitors, or γ-aminobutyric acid analogs (gabapentin or pregabalin) as first-line agents followed by opioids and topical treatments; again, these recommendations are targeted at adult patients and predominantly based on expert opinion. 114
MEN type 2B
MEN type 2B is a rare syndrome of autosomal dominant inheritance that accounts for 5% of all cases of MEN 2. The condition is associated with medullary thyroid carcinoma, pheochromocytoma, ganglioneuromatosis, as well as various skeletal and connective tissue abnormalities. 21 Patients with MEN 2B usually carry either an M918T or A883T mutation of the RET (rearranged during transfection) oncogene.21,115 Affected patients have a marfanoid habitus along with thick, fleshy everted lips and the appearance of eversion of the eyelids. 21 In addition, a high arched palate and firm pale neuromatous nodules on the tongue as well as atrophy of the fibula, pes cavus, scoliosis, and proximal or distal muscle wasting is reported. 22
This disorder may present with neuropathy manifesting as weakness of ankle dorsiflexion or occasionally of the intrinsic hand muscles. 25 Motor and sensory nerve conduction studies reveal mild impairments and chronic denervation on needle electromyography (EMG).23,25
Moderate loss of small- and large-diameter myelinated fibers is seen on sural nerve biopsy. Hyperplastic interlacing bands of Schwann cells and myelinated fibers that overlie the posterior columns of the spinal cord are reported in postmortem samples. 23 Involvement of autonomic nerves is also reported and leads to severe constipation or diarrhea.23,24
Owing to the high risk of malignancy, early diagnosis is vital, especially as medullary thyroid carcinoma is typically already established when the diagnosis is made.21,25 There is no specific therapy for the neuropathy beyond standard management and supportive interventions with rehabilitation and orthotics. It is not evident from the literature how treatment of the underlying neoplasias relates to the manifestation and or resolution of the neuropathy.
Vitamin deficiency states
Vitamin B1 (thiamine) deficiency
Thiamine deficiency occurs in infants who are breastfed by mothers with inadequate intake of thiamine or who receive low-thiamine-content formula,26,27 and in children who undergo medical or surgical procedures such as gastrointestinal resections, parenteral nutrition, and chemotherapy that result in inadequate thiamine uptake.29,116–118 The condition in infants is referred to as beriberi, while in children the manifestation falls under the term Wernicke’s encephalopathy. 29 Infantile encephalitic beriberi (IEBB) is a rare form of thiamine deficiency complicated by cardiac dysfunction, neck rigidity, and acute peripheral neuritis. 28 The life-threatening respiratory and encephalopathic symptoms can mimic Leigh syndrome. The childhood onset form, Wernicke’s encephalopathy, presents with the triad of ophthalmoplegia, ataxia, and confusion.29,116–118
The study by Ortigoza-Escobar et al., across 21 centers, reviewed the primary and secondary conditions leading to thiamine deficiency in infants and children. 29 The group identified 79 children with inherited thiamine defects associated with neurological manifestations. The majority were due to SLC19A3 disease (n = 70), the phenotype of which includes biotin thiamine responsive basal ganglia disease, Leigh’s syndrome (LS), infantile spasms with lactic acidosis, and Wernicke-like encephalopathy. 29 The remainder had TPK1 disease (n = 4) with a LS phenotype, and SLC25A19 disease (n = 5) associated with Amish microcephaly and bilateral striatal degeneration and progressive polyneuropathy. 29 Data was collected on 153 children (n = 65) and infants (n = 88) with secondary thiamine deficiency. Infantile and childhood thiamine deficiency is rare in resource-equipped settings. In low- and middle-income countries of Africa, staple foods of polished rice and incorrectly prepared cassava are deficient in thiamine, which leads to high risk for thiamine deficiency. 119
A predominantly sensory neuropathy with an additional acute motor axonopathy is detected on nerve conduction studies. 30 Large-fiber mainly axonal degeneration and subperineurial edema is typically found on biopsy. 31
Sustained clinical improvement is reported following thiamine supplementation in patients with secondary thiamine deficiency, with less than 20% suffering mortality or neurological sequelae, inclusive of their neuropathy.29,30
Vitamin B2 (riboflavin) deficiency
Adequate dietary intake of riboflavin is essential as it is not endogenously synthesized or stored in human tissues. Riboflavin is integral to a diverse range of metabolic pathways through its role as a precursor of essential cofactors flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD). These flavoproteins are important in chromatin remodeling, DNA repair, protein folding, and apoptosis. 32 Dietary deficiency, or ariboflavinosis, causes night blindness, cataracts, lethargy, anemia, poor growth, migraines, peripheral neuropathy, and dermatological symptoms. 32 Whilst unlikely with a normal, diet it is reported to occur during lactation, phototherapy in infants, in children with celiac disease, malignancies, or prescribed drugs that include phenothiazine-derived antipsychotic medications, the antimalarial drug quinacrine, phenobarbital, and the cancer chemotherapy agent adriamycin. 32
The Brown–Vialetto–Van Laere syndrome (BVVL) is a rare neurodegenerative disorder with progressive pontobulbar palsy, sensorineural hearing loss, optic atrophy, and muscle weakness related to an axonal sensorimotor neuropathy. 33 Without recognition and early intervention the condition is life-threatening owing to respiratory disease in early childhood. 33
BVVL is caused by homozygous or compound heterozygous mutations in the genes encoding the plasma membrane based riboflavin transporters leading to riboflavin transporter defect type 2 (RTD2) (SLC52A2 gene mutation) and RTD3 (SLC52A3 gene mutation).33,120,121 Children with RTD3 deficiency have sensorineural hearing loss, generalized lower limb predominant muscle weakness, bulbar weakness and tongue fasciculations, and respiratory weakness.25,122 RTD2 deficiency differs from the phenotype of RTD3, with sensory ataxia presenting in childhood as well as sensorineural hearing loss. Muscle weakness is limited to the upper limbs, leading to the ‘child-in-the-barrel’ phenotype. Serum acylcarnitines are elevated in about 50% of cases. 123 Neurophysiological studies typically show axonal sensory and motor changes with evidence of anterior horn dysfunction and chronic denervation. 32 Sural nerve biopsies confirm axonal neuropathy and degeneration preferentially affecting large-diameter myelinated fibers, which correlates with the sensory dysfunction in affected patients. 34 Muscle biopsy in isolated cases of children with RTD2 and RTD3 has shown evidence of mitochondrial dysfunction inclusive of complex II and III deficiency, and ragged red fibers consistent with mitochondrial myopathy.35,124
The pathogenesis of the hereditary disorder is being explored and may lead to novel therapies, especially related to the mitochondrial function.34,125 The ripple effect of the deficiency state leading to mitochondrial dysfunction is most likely a secondary effect rather than part of a primary mitochondrial disorder. The manifestation of features compatible with mitochondrial dysfunction highlights the importance of including RTD in the differential diagnosis. 124
Early recognition of RTD is important, as treatment with high-dose riboflavin (30–80 mg/kg/day) is well tolerated and slows or reverses the neuropathy and hearing loss. 35
Vitamin B6 (pyridoxine) deficiency
Vitamin B6, a water-soluble vitamin found in many standard dietary products, is a coenzyme for many reactions and involved in neuronal signaling through the synthesis of neurotransmitters. 39 Deficiency states are associated with cardiovascular dysfunction and polyneuropathy. 39 Ascending acute (<4 weeks) or subacute (12 weeks) numbness, with neuropathic pain, balance impediment, and weakness, is reported. 38 Neurophysiological studies are markedly abnormal, showing length-dependent acute axonal sensorimotor polyneuropathy with denervation, or a sensory neuropathy. 38
Sural nerve biopsy shows a severe active axonal neuropathy with multifocal loss of large and small myelinated fibers, with scant epineurial vascular proliferation and inflammation. 38 This loss of small myelinated fibers is a complication as highlighted by an adult patient with additional autonomic dysfunction. 126
Isoniazid-induced painful sensory neuropathy is relatively common in adults, and has also been reported in a few childhood cases.36,37 Supplemental pyridoxine (5–10 mg/day) is recommended in HIV-positive or malnourished children receiving treatment for tuberculosis to avoid this complication. 127
Pyridoxine is converted into its active form, pyridoxal phosphate. High levels of pyridoxine are thought to inhibit pyridoxal-phosphate-dependent enzyme. Paradoxically this means that supplementation with vitamin B6, usually in doses above 50 mg day, is associated with the development of a painful sensory neuropathy.39,40 To date this has only been reported in adults.128,129 The toxicity of vitamin B6 is not only dose determined, but related to the vitamer in which it is taken. There is a proposal that pyridoxine should be replaced by pyridoxal or pyridoxal phosphate when vitamin B6 supplements are required. 39
Vitamin B12 deficiency
Childhood onset of vitamin B12 deficiency anemia owing to impaired absorption of cobalamin, or poor dietary intake, is well known, but more often with myelopathic rather than neuropathic symptoms. 41 Symptomatic vitamin B12 deficient-neuropathy is more often seen in resource-poor settings.130–132 Early recognition and intervention are important to prevent irreversible nerve injury. 133 A study of 66 adolescents and adults with a vitamin B12 deficient neurological syndrome found that 69.7% had clinical features of neuropathy and 54.5% had abnormal nerve conduction studies, most of which had mixed features of axonal and demyelinating disease. 42 Nerve biopsy in the early stages was consistent with acute axonal degeneration, and in later stages showed a chronic axonopathy with secondary demyelination.
With appropriate supplementation and management of underlying etiologies, nerve conduction parameters, and clinical findings improve within 6 months. 42
Vitamin E deficiency
Children with protein-energy malnutrition, long-standing obstructive liver disease, chronic intestinal malabsorption, and cystic fibrosis are at risk of a progressive neurological syndrome owing to defective or inadequate vitamin E absorption.43,134–136 Vitamin E deficiency is also a major cause of the polyneuropathies that occur in children with abetalipoproteinemia or hypobetalipoproteinemia. 137 These conditions are autosomal recessive disorders of the synthesis of the beta-lipoprotein owing to mutations of the MTTP (microsomal triglyceride transfer protein) or APOB genes, respectively.138,139 Affected children develop nystagmus, gaze palsies (especially vertical gaze), retinitis pigmentosa, night blindness, ataxia, areflexia as well as impaired position and vibration awareness.43,44 Neurological features may be evident by 2 years of age, with a third of affected children being symptomatic by 10 years. Intellectual disability affects the same proportion. The clinical phenotype can mimic Friedreich ataxia and other similar primary spinocerebellar ataxia diseases. 47 Abetalipoproteinemia is treatable through the supplementation of fat-soluble vitamins and dietary modification. 139
Vitamin E malabsorption can also occur due to familial isolated vitamin E deficiency caused by mutations in the alpha-tocopherol transfer protein gene (TTP1) and referred to as ataxia with vitamin E deficiency (AVED)44,140 Most patients are from the Mediterranean region. 47 The antioxidant properties of vitamin E have a role in modulating glutamate neurotoxicity. In this genetic condition, failure of uptake of vitamin E into plasma low-density lipoproteins impairs normal recycling of vitamin E. Clinical features of AVED include dysarthria, progressive ataxia, weakness, loss of deep tendon reflexes, and impaired proprioception, with absent or very low serum vitamin E levels in the absence of hypolipidemia or fat malabsorption. Symptoms usually commence between 3 and 13 years of age (range 2–37 years). 47 Patients with AVED do not usually have retinopathy or ophthalmoplegia.44,141 Upgoing plantar responses, tremor (especially head tremor), and dystonia of movements may occur.45,47
Somatosensory evoked potential studies confirm the abnormal posterior column function.45,46 A sensory neuronopathy with normal motor conduction and absent or markedly reduced sensory nerve action potentials (SNAPs) is typically recorded on neurophysiologic studies.47,48 Nerve biopsy typically shows axon los.45,47 Clinical resolution, as well as improvement in abnormal sensory conduction and evoked potentials, is reported with dietary vitamin E supplementation (up to 800 mg/day). 49 Intervention early in the disease course generally leads to better outcomes, but some patients are resistant even with timelysupplementation. 47
Malignancies and reticuloses
Children with cancers are at risk of PNS dysfunction, for multiple reasons including direct invasion, paraneoplastic syndromes, nerve compression, the side-effects of chemotherapy, infection, and the sequelae of poor nutrition.50,65,142,143 However, once drug side-effects and local malignant infiltration are excluded, neuropathies are a relatively rare complication of childhood malignancies. 143 In 49 childhood malignancy patients who were not managed with vincristine, none of the group had evidence of clinical or electrophysiological features of neuropathy. 144 From the same study, in 47 children treated with vincristine there was a high rate of absent deep tendon reflexes and abnormal sensory conduction velocities, but in most cases children were not clinically compromised. 144
Lymphoma
Peripheral neuropathy manifests in about 5% of adults with lymphoma, but is much rarer in childhood. 50 A sensory neuropathy and a sensorimotor neuropathy occur as the two main carcinomatous neuropathies. Genetic markers of increased predisposition to development of vincristine-induced peripheral neuropathy (VIPN) in children with acute lymphoblastic leukemia (ALL) have recently been identified, and may offer a means to tailoring therapy to avoid this disease complication. 145
Pure sensory neuropathy is more likely to be seen in association with carcinoma rather than lymphoma. 144 The symptoms of pins and needles, altered unpleasant skin sensation, discomfort, and proprioceptive ataxia are similar to those seen in carcinoma. Small-fiber neuropathy is reported in a third of children with ALL who completed treatment protocols, which include vincristine. 52 For patients with combined sensory and motor neuropathy, acute polyneuropathy of the acute inflammatory polyradiculoneuropathy (AIDP) phenotype is reported in patients with lymphoma, especially Hodgkin’s disease. Differentiating between AIDP and vincristine toxicity can be challenging in children with ALL, non-Hodgkin’s and Hodgkin’s lymphoma57,146,147 who present with a mainly lower limb sensory and motor neuropathy.57,146 Early in the course of the condition, F waves may be absent in children with AIDP, and these children can have a good response to intravenous immunoglobulin (IVIG). 146 A more chronic demyelinating or axonal neuropathy also occurs in pediatric lymphoma. 51 Nerve roots and peripheral nerve malignant invasion occurs more often in lymphoma than in carcinoma. In most cases management of associated neuropathy is symptomatic and supportive. Glutamine supplementation is reported to be well tolerated with improvements in sensory function and overall quality of life. 53 Future research is needed to establish if glutamine should form part of the care for patients with vincristine-related sensory neuropathy.
Paraneoplastic neuropathies
About 1% of cancer sufferers are affected by paraneoplastic neurological syndromes. Adults tend to develop peripheral neuropathies mediated by antibodies to intracellular (Hu-D and CV2-CRMP5) or cell membrane antigens (voltage-gated calcium channel, LG1 and CASPR2 proteins).65,142,148 Children more typically develop autoimmune encephalitis rather than peripheral nerve dysfunction, 149 but rare cases of paraneoplastic neuropathy are reported.54–56 A 13-year-old boy with Hodgkin’s disease was reported with an acute polyneuropathy and autoimmune hemolytic anemia. 54 Axonal degeneration in the nerves and dorsal funiculus was identified on pathological assessment, and these nerves had no evidence of metastatic spread or cellular inflammation. Occasional cases of atypical pediatric AIDP and CIDP represent a paraneoplastic complication of lymphoreticular malignancies.55–58 Improvement is reported with IVIG and supportive care. 57 Development of peripheral neuropathy in a patient with non-Hodgkin’s malignant lymphoma (NHML) is an indication for intensification of chemotherapy. This approach can lead to significant regression of neuropathy. 58
Graft-versus-host disease
Allogeneic hematopoietic stem cell transplantation in children and adults can be complicated by acute immune-mediated neuropathies.59–63 In relation to graft-versus-host disease (GVHD), all subtypes of Guillain–Barré syndrome can occur, often after intercurrent infections. 60 Affected patients typically have a good response to intervention with IVIG. A 7 month old girl post-bone marrow transplantation who developed severe GVHD, had her course further complicated by a chronic inflammatory demyelinating neuropathy. 64 Whilst a graft-versus-host response places children at risk of peripheral neuropathy, other direct or indirect causes (for example, toxins, infections, and paraneoplastic processes), should also be considered.150,151 For example thalidomide is often used in the management of graft-versus-host response, and peripheral neuropathies can occur with use of this agent. 152
Neurofibromatosis
Neurofibromatosis type 1 (NF1), an autosomal dominant condition, may be complicated by peripheral nerve tumors [spinal neurofibromas, plexiform neurofibromas, and malignant peripheral nerve sheath tumors (MPNST)]. 153 NF2, which is not as prevalent as NF1, less commonly manifests in childhood, but can also be associated with neuropathies. Sarcomatous malignant change occurs in some 8–13% of affected people with peripheral nerve sheath tumors, with a poor prognosis.67,68 The histological appearance is illustrated by Figures 1 and 2. Mutations in the p53 and INK4a genes, and aberrant signaling in the Notch pathway are factors leading to the development of MPNST.65,66 Of 123 Chinese children with NF1, three patients (2.4%) developed had MPNST. 66 Severe pain and rapid growth of the soft-tissue lesion are markers of malignant change. 65 Early detection of this complication may be possible with serum markers. 68 High-resolution ultrasonography may be of value in detecting peripheral nerve involvement in NF1 and NF2.154,155 There is no therapy to attenuate or prevent neuropathic symptoms of NF1, 67 but MEK and mTOR inhibitors have been proposed as possible interventions for tumor-associated neuropathic pain. 67 Surgical resection has been effective in some case reports where neuropathy is not associated with malignant transformation. 156 The use of high-resolution intraoperative ultrasound and contrast-enhanced ultrasound is reported to improve surgical outcomes. 157 Collaborative groups are developing research to aid establishment of effective management protocols. 158
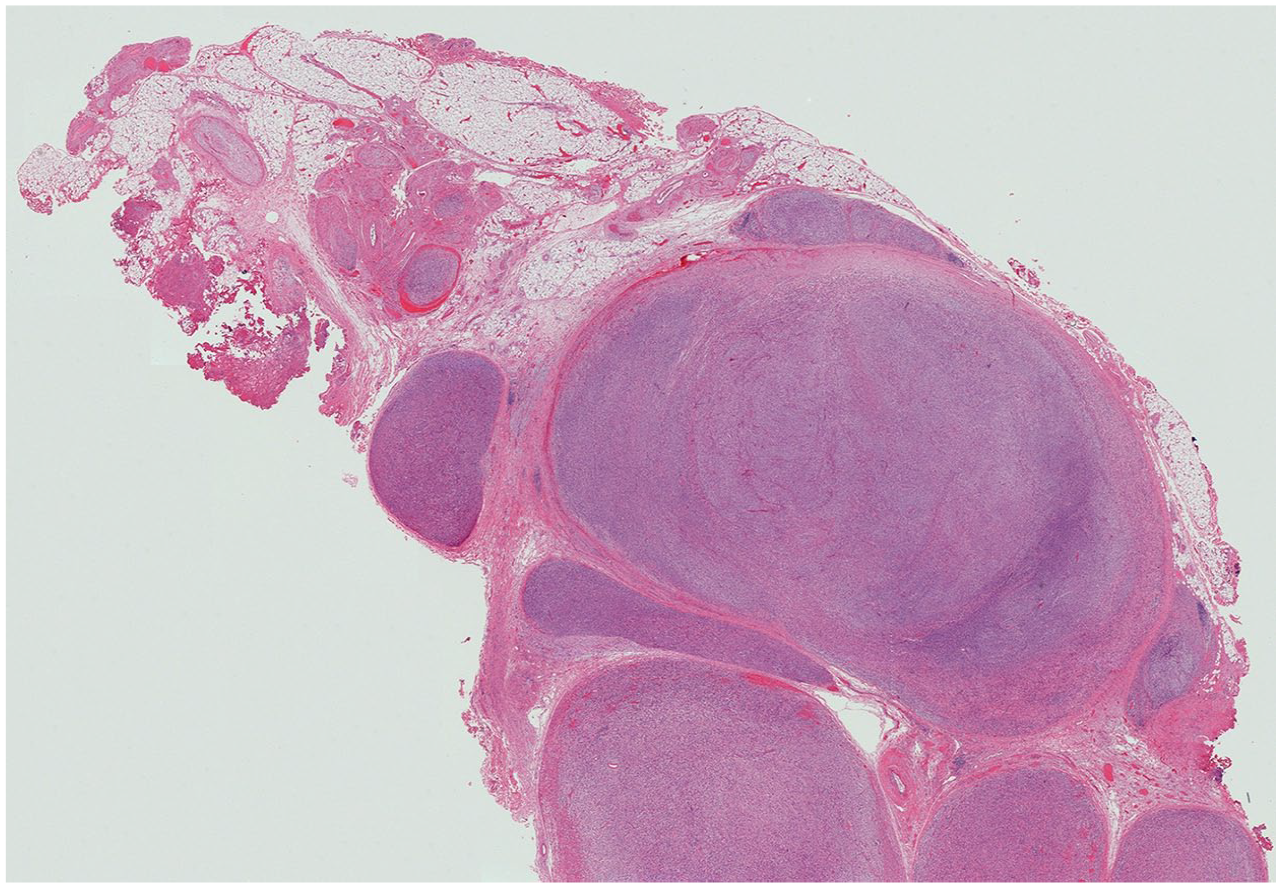
Tumor is seen arising within a plexiform neurofibroma in a patient with of Neurofibromatosis type 1 (NF1) on low-power montage. (Image supplied by Associate Professor Duncan McGregor, Department Head of Anatomical Pathology, The Royal Children’s Hospital, Melbourne, Australia)

The same patient with NF-1 as in Figure 1, in this view tumour is seen arising within a plexiform neurofibroma at high magnification illustrating the densely cellular spindle cell sarcoma is visible. (Image supplied by Associate Professor Duncan McGregor, Department Head of Anatomical Pathology, The Royal Children’s Hospital, Melbourne, Australia)
Sickle cell disease
Children with sickle cell disease, a condition prevalent throughout the African population, are predisposed to the development of a demyelinating peripheral neuropathy in the setting of lead intoxication. 69 In many resource-poor countries, lead intoxication remains a risk; this is an avoidable exacerbating factor to exclude in children presenting with neuropathy who have sickle cell disease. Chelation therapy was reported to result in improvement of the motor function in the two reported children over a period of months. 69 Sensory neuropathy in patients with sickle cell disease is reported more outside the childhood age range. 159
Connective tissue disorders
Rheumatoid arthritis
Adults with rheumatoid arthritis are more likely to suffer polyneuropathy and entrapment neuropathies than children. 70 Carpal tunnel syndrome is occasionally seen in children with juvenile rheumatoid arthritis.160,161 Delayed nerve conduction in single nerves was reported in 4 children from a cohort of 99 with juvenile rheumatoid arthritis. 162 Clinically there was no evidence of mononeuropathy, and conduction in the other nerves was normal. 162 A 14-year-old boy with established juvenile rheumatoid arthritis, over 4 months developed muscle weakness and reduced deep tendon reflexes, he had denervation on EMG, but normal motor nerve conduction studies. 8 Symptoms, inclusive of neuropathic pain, experienced by patients with rheumatoid arthritis may be mild or unusual in childhood, but become more pronounced with time, such that 96% of adults have symptomatic neuropathy. 71 Children benefit from optimal care of their underlying rheumatoid arthritis and symptomatic relief of neuropathy where required. 163
Systemic lupus erythematosus and other vasculitic disorders
Symptomatic peripheral nerve disease is uncommon in children with systemic lupus erythematosus (SLE), however subclinical nerve involvement is reported in up to 15% of affected children.164–169 Whilst children may present with SLE owing to complications of a neuropathy, for example foot drop or pain, 170 most children with SLE who develop neuropathy do this between 1 and 5 years after the diagnosis. 165 The neuropsychiatric manifestations, which are common in the setting of SLE, can further complicate recognition of a neuropathy. 170 Mononeuritis multiplex, an acute sensorimotor neuropathy similar to GBS, and a distal sensory neuropathy are the three main types of nerve disease to occur. Nerve conduction studies usually show sensory and motor axonal pathology, which may be slowly progressive or may wax and wane over time, generally worsening with age.72,73 Axonal degeneration affecting myelinated and unmyelinated fibers is found on sural nerve samples, in addition to vasculitis in most cases.74,75 Pathogenetic mechanisms suggested for the peripheral neuropathy in SLE include vasculitis of the vasa nervorum, endoneurial immune complex deposition and antiphospholipid antibody-mediated damage to components of neural tissue.75,164,165,168,171 The vasculitis leads to ischemic infarction of blood vessels, which results in Wallerian degeneration of nerve fibers.172,173 Treatment may include immunomodulation and IVIG. 76 Children with SLE should undergo regular nerve conduction studies to monitor for development of subclinical neuropathy.164,170
Vasculitic mononeuritis multiplex has been described in children with SLE, Churg–Strauss syndrome, and polyarteritis nodosa.174–180 Sjögren’s syndrome is an autoimmune exocrinopathy primarily affecting the salivary and lacrimal glands. This condition is also associated with development of a sensory neuropathy possibly related to vasculitis or ganglionitis of the dorsal roots. 181 Occasional pediatric cases are reported. 181 Immunosuppression and immunomodulation are part of standard care. Children with polyarteritis need aggressive immunosuppression, especially in the setting of progressive gangrene related to the sensory vasculitic neuropathy. 182 There are conflicting studies in relation to the role of the tumor necrosis factor-alpha agent, etanercept.183,184
Miscellaneous systemic disorders
Chronic idiopathic intestinal pseudo-obstruction
Intestinal pseudo-obstruction can affect all ages from the neonates through to adults. A motor neuropathy may occur related to neuronal dysplasia in the myenteric plexus, as well as in association with Duchenne muscular dystrophy, myotonic dystrophy, mitochondrial cytopathy, myxedema, porphyria, scleroderma, amyloidosis, and Chagas disease.77,185–187 Familial visceral neuropathy is the hereditary form of the disorder. 77 Nonintestinal abnormalities are reported, including pupillary abnormalities.188–190 There is overlap with the features of the condition familial visceral myopathy.77,185 Demyelinating changes are typically recorded on nerve conduction studies, although axonal pathology is also reported. 77 The mode of inheritance is not defined. 77
Waardenburg–Shah syndrome also combines bowel dysmotility and peripheral neuropathy; this subtype is allelic to Waardenburg syndrome type 4 due to recessive mutations in the SOX10 gene.78,79 This neurocristopathy presents early in life with Waardenburg syndrome, central dysmyelination, Hirschsprung disease, and a hypomyelinating peripheral neuropathy.78–85 Hypoplasia and markedly reduced axonal fibers were detected on sural nerve biopsy of an infant with Waardenburg–Shah syndrome. 82
Whilst peripheral neuropathy is common in the setting of mitochondrial disease, especially mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS) and myoclonic epilepsy with ragged-red fibers (MERRF), mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) invariably causes peripheral neuropathy. 191 MNGIE is a rare multisystemic recessive disorder which is complicated by progressive external ophthalmoplegia, gastrointestinal impaired motility, extreme weight loss, muscle wasting, peripheral neuropathy and leukoencephalopathy, due to mutations in the TMMP gene, encoding for thymidine phosphorylasethymin phosphylase. 192 Onset of the condition is in childhood or adolescence. 193 The peripheral neuropathy is usually demyelinating but mixed axonal/demyelinating or predominantly axonal cases are reported. 192 Delay in diagnosis of MNGIE is common. 192 Allogeneic haemopoietic stem cell transplantation may improve the neuropathy as well as improving body mass index and gastrointestinal manifestations. 194
Celiac disease is a gluten-sensitive enteropathy complicated by an axonal or demyelinating sensory or sensory and motor neuropathy in some patients.86–88 Recurrent Guillain–Barré syndrome occurred in a single patient which was considered due to molecular mimicry.195,196 A 3-year-old child who had celiac disease developed an evolving polyneuropathy that was unresponsive to a gluten-free diet, vitamin E and folic acid supplementation, and on sural nerve biopsy had marked reduction of myelinated fibers and no evidence of regeneration. 89 A Wallarian degeneration mechanism was supported by the distal involvement and the nerve biopsy findings, further the failure to respond to a gluten-free diet supported that direct toxicity of gliadin had not occured. 89 Neurophysiologic screening is not recommended in asymptomatic patients with celiac disease. 90 A large Swedish population study, however, concluded that patients with idiopathic immune peripheral neuropathies should be screened for celiac disease. 91
Sarcoidosis
Peripheral neuropathy involvement in sarcoidosis may be isolated or part of the overall systemic disease complications. 93 Adults, in particular, are at risk of facial nerve palsy, which is the most common neurological manifestation of the condition. 93 Generalized neuropathies are usually asymmetrical and present as mononeuritis multiplex or Guillain–Barré syndrome. 93 Sensory loss, with altered skin sensation or pain, over large areas of the torso, can also occur. 94
Cranial neuropathy in addition to generalized chronic neuropathy is very supportive of a diagnosis of sarcoidosis. This is typically in the setting of a patient with hilar lymphadenopathy, uveitis, parotitis, and erythema nodosum. 92 Overall peripheral nerve disease in children is rare but has been reported as young as 13 years of age, with two cases presenting with Guillain–Barré syndrome.92,197 Neurosarcoidosis was reported in 53 children who most often manifested with cranial neuropathy (in 21% of cases). 96
Nerve conduction studies can be normal or detect mild slowing of conduction. 94 Muscle or sural nerve biopsy may identify sarcoid granulomas. 95 Mild pleocytosis and raised total protein can occur in cerebrospinal fluid. 94
Whilst the long-term outcome of sarcoid neuropathy is better than that of central nervous system sarcoidosis, the overall prognosis cannot be predicted. Although corticosteroids are used, the evidence to support this intervention is not established. 96
Conclusion
This review provides an overview of systemic conditions that can be associated with PNS dysfunction. Some neuropathies associated with systemic disease, such as CIP and autonomic dysfunction with diabetes mellitus, can have a profound clinical effect and compromise. In other settings, screening for evidence of nerve dysfunction can be an indicator of chronic poor disease control. In some conditions, such as celiac disease, peripheral neuropathy may be the presenting feature warranting active exclusion of the systemic condition. In all cases early recognition and symptomatic care of neuropathy is important for mobility and pain control. In many conditions, neuropathy will improve in tandem with the optimal management of the underlying disease. In conclusion, peripheral neuropathies can be associated with a diverse range of medical conditions and unless actively considered may not be recognized and, thus, inadequately managed.
Footnotes
Acknowledgements
Figures 1 and 2 were provided by Associate Professor Duncan McGregor, Department Head of Anatomical Pathology, The Royal Children’s Hospital, Melbourne, Australia.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
The authors declare that there is no conflict of interest.