
Abstract
Objectives:
Approximately one in two patients with multiple sclerosis (MS) suffer from comorbid depression. The primary objective of this study was to evaluate the safety and tolerability of fingolimod and antidepressant combination in relapsing–remitting MS patients with mild-to-moderate depression. Efficacy outcome variables were quality of life (QoL), fatigue, disability and depression.
Methods:
Patients received open-label fingolimod 0.5 mg over 2 weeks, followed by fingolimod plus citalopram (40 mg), fluoxetine (40 mg) or venlafaxine (150 mg) over 16 weeks. The antidepressant was selected at the physician’s discretion.
Results:
In total, 54 patients were recruited at 25 centres across Germany. No new safety signals (including cardiac) emerged compared with previous clinical studies. Adverse events (mostly mild-to-moderate) were reported in 43 patients. A total of three patients had serious adverse events and 10 discontinued the study. QoL (mean [95% confidence interval]) improved by 2.2 (−3.3, −1.2; Patient Reported Indices for MS questionnaire), fatigue by 8.2 (−13.1, −3.3; modified Fatigue Impact Scale) and depression by 6.3 (−8.4, −4.2; Hamilton Depression Scale) points. However, the results must be interpreted cautiously owing to limited patient numbers.
Conclusions:
Combination of fingolimod with antidepressant medication showed no unexpected safety signals. Patient-reported outcomes (QoL, disability, fatigue and depression) remained stable or improved.
Introduction
Multiple sclerosis (MS) is a chronic, demyelinating, immune-mediated disease of the central nervous system (CNS). Fingolimod (Gilenya®, FTY720; Novartis Pharma AG, Basel, Switzerland) is a sphingosine-1-phosphate (S1P) receptor modulator approved in several countries as a 0.5 mg once-daily oral therapy for relapsing MS. The safety and efficacy of fingolimod in relapsing–remitting multiple sclerosis (RRMS) is well characterized [Cohen et al. 2010; Comi et al. 2010; Kappos et al. 2010; Calabresi et al. 2014]. As of August 2015, approximately 132,000 MS patients have been treated with fingolimod in both clinical trials and postmarketing settings. The total patient exposure is approximately 265,000 patient-years (Q3 Novartis Pharmaceuticals Interim Financial Report, August, 2015).
Depression is frequently reported in MS patients with a lifetime risk of 40–60% [Vattakatuchery et al. 2011]. Several studies have indicated up to three times higher prevalence of depression in MS patients compared with the general population [Patten et al. 2003; Mohr et al. 2006; Beiske et al. 2008; Ziemssen, 2009]. Depression in MS patients leads to impairment of various components of quality of life (QoL), and if untreated, can lead to suicidal ideation and poor adherence to MS treatment [Mohr et al. 2006; Beiske et al. 2008; Ziemssen, 2009]. Despite the high burden of depression in MS patients, the condition is still under-recognized and under-treated.
Literature on the use of antidepressants in MS patients is limited to two double-blind (N = 28 and N = 42), placebo-controlled studies and a few open-label studies. These studies have indicated favourable results with respect to depression-related symptoms; however, the results were based on limited sample sizes [Schiffer and Wineman, 1990; Ehde et al. 2008; Solaro et al. 2013]. There is one study that evaluated the efficacy of an antidepressant (fluvoxamine) in MS patients with major depression who were receiving a stable dose of MS disease-modifying therapy (DMT; interferon beta-1b) [Benedetti et al. 2004].
The Reduce dEpression with Gilenya And SNRI/SSRI antIdepressant in MS patieNts (REGAIN) study was conducted primarily to assess the safety and tolerability profiles of a combination of fingolimod with widely-prescribed selective serotonin reuptake inhibitors (SSRIs; citalopram and fluoxetine) or serotonin and noradrenaline reuptake inhibitor (SNRI; venlafaxine) in RRMS patients with comorbid mild-to-moderate depression in Germany. Exploratory MS- and depression-related efficacy parameters were also assessed, including fatigue, QoL, treatment satisfaction and disability.
Methods
Study design
REGAIN was a 21-week, multicentre, open-label study [ClinicalTrials.gov identifier: NCT01436643], comprising a screening phase (up to 3 weeks), an open-label treatment phase, during which patients received fingolimod 0.5 mg for at least 2 weeks, followed by a 16 weeks core phase during which patients received fingolimod in combination with citalopram, fluoxetine or venlafaxine (Figure 1).

Study design.
To assure adequate sample size in each treatment arm, we only selected four of the most commonly used antidepressants.
The antidepressant was assigned according to the investigator’s discretion and not by randomization. The antidepressant was titrated over 7 to 14 days, from an initial dose of 20 mg citalopram, 20 mg fluoxetine or 75 mg venlafaxine up to a final dose of 40 mg citalopram, 40 mg fluoxetine or 150 mg venlafaxine.
During the study period, the Committee for Medicinal Products for Human Use and the European Medicines Agency reviewed the benefit–risk profile of fingolimod and recommended a label update for Gilenya® to include first dose monitoring, and the protocol was amended accordingly. In addition, the ethics committee advised additional weekly electrocardiogram (ECG) recordings in the citalopram arm after two ‘Dear Health Care Professional (DHCP) Letters’ were published for citalopram and fingolimod. Because this monitoring would have extremely minimized the willingness of investigators for the combination treatment, recruitment in the citalopram arm was stopped. Patients who had already received citalopram continued the treatment as planned.
Patients
The study included patients with RRMS (2010 revised McDonald criteria [Polman et al. 2011]) aged between 18–65 years and diagnosed with comorbid depression (according to the International Statistical Classification of Diseases and Related Health Problems [ICD]-10 [World Health Organization, 2016]) of mild-to-moderate intensity (Beck Depression Inventory second edition [BDI-II] score of 14–28) [Beck et al. 1996]. Key inclusion and exclusion criteria are provided in Table 1.
Inclusion and exclusion criteria.

BDI-II, Beck Depression Inventory second edition; DMT, disease-modifying therapy; EDSS, Expanded Disability Status Scale; Gd+, gadolinium enhancing; IgG, immunoglobulin G; MS, multiple sclerosis; RRMS, relapsing–remitting multiple sclerosis.
The study was conducted in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice and the Declaration of Helsinki. The protocol was approved by the Bayerische Landesärztekammer ethics committee, Germany. Each patient provided written informed consent before enrolment.
Safety assessment
The primary outcome variables included severity and frequency of adverse events (AEs). Safety assessments performed at each study visit, included reporting of AEs, serious adverse events (SAEs), vital signs, physical and neurological examinations and laboratory examinations. ECGs were recorded in a subset of 29 patients at each visit after visit 3 (after uptitration of the antidepressant at visit 3.1).
Efficacy assessments
Several exploratory efficacy variables were assessed. The impact of treatment on QoL and daily activities was assessed using the Patient-Reported Indices for Multiple Sclerosis (PRIMUS) scale [Doward et al. 2009]. The severity of the depression-related symptoms was assessed using the 21-item Hamilton Scale for Depression (HAM-D21) [Hamilton, 1960] and BDI-II [Beck et al. 1996]. The impact of treatment on fatigue was assessed using the modified Fatigue Impact Scale (mFIS) [Fisk et al. 1994]. MS-related disability was assessed using the Expanded Disability Status Scale (EDSS) [Kurtzke, 1983]. For all five scales, higher scores indicated greater impairment of symptoms. Treatment satisfaction with medication was assessed using the Treatment Satisfaction Questionnaire for Medication (TSQM-9) [Bharmal et al. 2009] in which higher scores signify higher satisfaction. Resource utilization data for MS and for depression were collected from patients via questionnaires referring to the past 10 weeks, if not stated otherwise. All exploratory variables were measured at baseline and week 16 except EDSS, which was measured at screening and week 16.
Statistical analysis
Approximately 250 patients would have ensured a representative sample of antidepressant usage for the study. The first patient was recruited in November 2011 but the necessary patient number was not reached by March 2013, even though recruitment had already been extended 30 weeks past the planned recruitment period. After discussions and feedback from the investigators it was considered very unlikely that the number of planned patients would be achieved, even after a considerable amount of time.
At the time of the decision to terminate the study early at the end of April 2013 due to slow patient enrolment, 61 out of the 250 anticipated patients had entered the study.
Overall, 54 patients received at least one dose of fingolimod and were included in the safety set. As the primary outcome was safety and the study was terminated prematurely, all analyses were conducted in the safety set only. Summary statistics (n, mean, median, standard error [SE] and confidence interval [CI]) were provided for all exploratory endpoints. The change from baseline to week 16 (mean, SE and CI) was reported for all exploratory variables. Changes in exploratory endpoints were tested by a one-sample Student’s t-test, with p < 0.05 as the level of significance.
Results
Patient disposition, demographics and baseline characteristics
This study was conducted in 54 patients at 25 centres across Germany from November 2011 to September 2013. All patients were white, and the majority were women (n = 44; 82%). The mean ± standard deviation (SD) age was 41.8 ± 9.9 years. The mean duration of MS since diagnosis was 8.9 ± 7.4 years, and the mean duration between first MS symptoms and MS diagnosis was 2.5 ± 4.0 years (Table 2).
Demographics, disposition and baseline characteristics by treatment and MS disease characteristics (all patients).
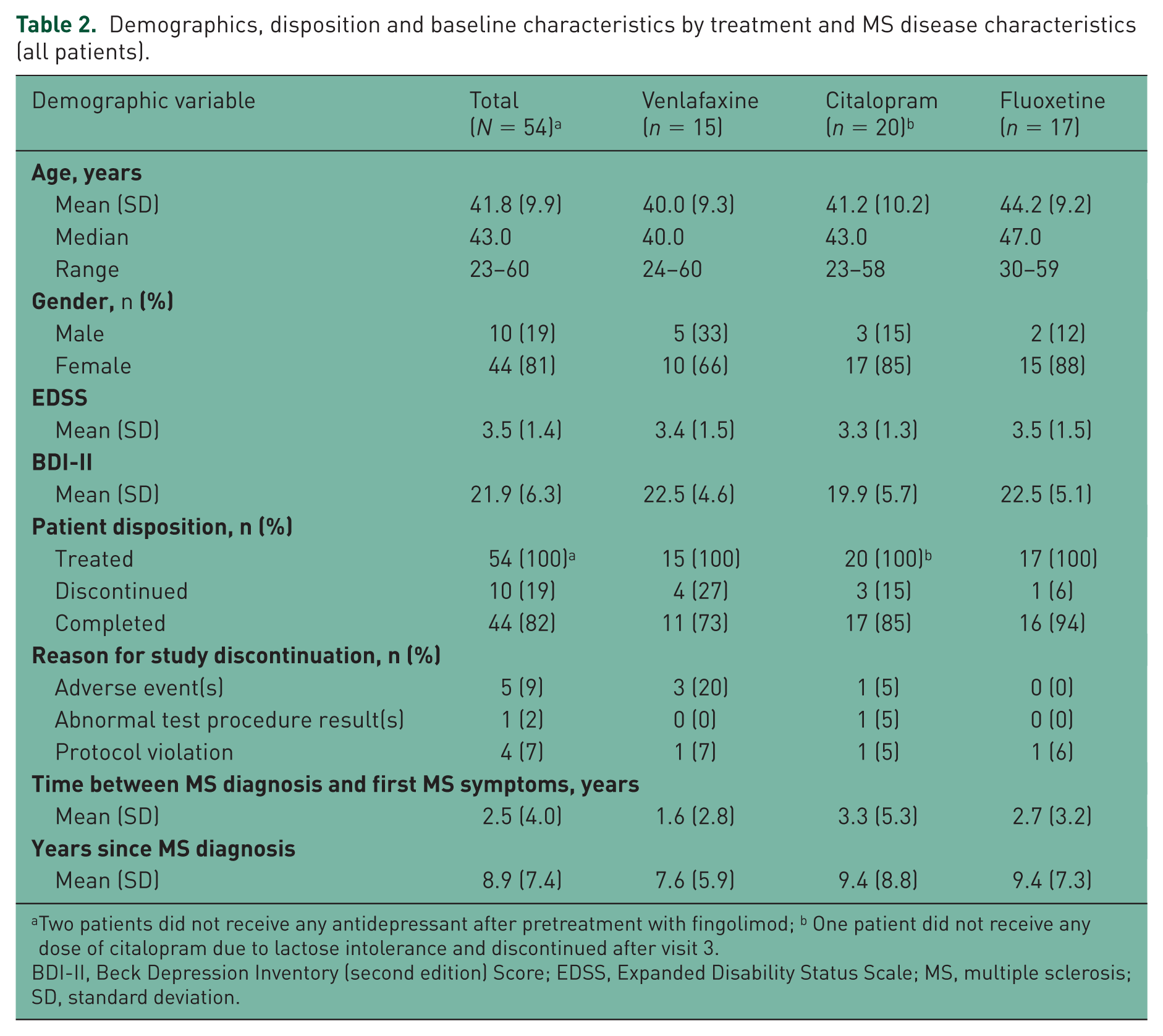
Two patients did not receive any antidepressant after pretreatment with fingolimod; b One patient did not receive any dose of citalopram due to lactose intolerance and discontinued after visit 3.
BDI-II, Beck Depression Inventory (second edition) Score; EDSS, Expanded Disability Status Scale; MS, multiple sclerosis; SD, standard deviation.
With the exception of three patients, all subjects enrolled had mild or moderate depression, as assessed by the BDI-II score (14–28). In two patients, the BDI-II was below 14 at screening, visit 2 (start of fingolimod) and visit 3 (start of antidepressant) (BDI-II patient 1: 13/11/11; patient 2 12/7/7, respectively). In a third patient BDI-II scores were below 14 at visit 1 and 2, but above 14, when antidepressant treatment was started (BDI-II 9/10/15, respectively).
Overall, 51 of the 54 patients received antidepressants: citalopram (n = 19), fluoxetine (n = 17) and venlafaxine (n = 15). A total of two patients discontinued prior to the first antidepressant intake: one due to an SAE (cerebral haemorrhage) and the other due to an out-of-range BDI-II score after visit 2, and were not included in the analysis. One patient assigned to receive citalopram did not take the citalopram film-coated tablet due to lactose intolerance. This patient was discontinued from the study but was included in the analysis. Overall, of the 54 patients included, 10 discontinued the study early due to SAEs or AEs (n = 5; 9%), protocol deviations (n = 4; 7%) and abnormal test procedure results (n = 1; 2%; Table 2). Overall, seven patients discontinued the SNRI/SSRI or changed dose due to AEs; two in the citalopram group and five in the venlafaxine group.
Study drug exposure
The mean ± SD duration of exposure to fingolimod was 108 ± 38 (median, 126; range, 11–143) days and that for citalopram, fluoxetine and venlafaxine was 97 ± 33, 107 ± 25 and 89 ± 47 days respectively. The mean daily dose of the antidepressants was: citalopram, 35 ± 6 mg; fluoxetine, 36 ± 4 mg and venlafaxine, 126 ± 27 mg.
Safety results
Of the total 54 patients, 43 (80%) experienced at least one AE; most AEs were mild or moderate in severity. In total, 13 patients had AEs with a suspected relationship to fingolimod and 20 to the antidepressant. No deaths were reported during the study.
SAEs were reported in three patients (cerebral haemorrhage, MS relapse and suicidal ideation). Only cerebral haemorrhage was suspected to be related to fingolimod, and the study drug was discontinued temporarily. This patient had a history of fall/trauma because of gait disturbance as a consequence of MS but had no history of cerebrovascular events, cardiac disorders, hypertension, migraine or diabetes mellitus. The patient recovered completely. In the investigator’s opinion, trauma due to gait disturbance could also be a possible contributory factor for this event. The patient with MS relapse was hospitalized and treated with medications including corticosteroids. The patient with suicidal ideation permanently discontinued the study due to the event.
Before starting antidepressant therapy, 33 AEs occurred in 24 patients; the most common were lymphopenia (n = 6), nasopharyngitis (n = 2), MS relapse (n = 2) and aggression (n = 2). Of note, all six cases of lymphopenia were reported from one centre and at the time of reporting none of these patients had a lymphocyte count lower than 300/mm3.
After starting antidepressant therapy, 94 AEs occurred in 36 patients; the most common were nausea (n = 11), MS relapse (n = 5), headache (n = 4) and nasopharyngitis (n = 4) (Table 3). Clinically significant abnormal laboratory results were documented in 10 (19%) patients: potassium > 6 mmol/l (n = 6), haemoglobin < 10 g/dl (n = 3) and bilirubin > 2 mg/dl (n = 1). The one cardiac event (tachycardia) reported in the venlafaxine group was considered to be causally related to venlafaxine. No clinically significant changes were observed in the atrioventricular conduction, QTc interval (Table 4), vital signs or blood pressure.
Number of patients with adverse events after first intake of antidepressant by preferred term (at least 10% incidence in total; safety set).

AEs, adverse events.
Note: One patient did not receive citalopram film-coated tablet due to lactose intolerance. This patient discontinued from the study but was included in the analysis.
ECG findings by treatment (ECG group; N = 29).

A total of five patients in the ECG group were treated with fingolimod before visit 2; three patients in the venlafaxine and two patients in the fluoxetine group; *N = numbers of patients with ECG available at respective visit/interval
ECG, electrocardiogram.
Efficacy results
The efficacy analysis indicated favourable results for most of the parameters at the end of study compared with baseline.
On the PRIMUS scale, the mean change from baseline with respect to QoL (n = 51) was −2.2 (95% CI: −3.3, −1.2), indicating an improvement, whereas the daily activities score remained stable with a mean change of 0.3 (95% CI: −1.2, 1.8; Figure 2). Treatment effect on depression was measured using HAM-D21 and BDI-II. On both the scales patients indicated improvements from baseline during the study. HAM-D21 score (n = 49) improved on average by −6.3 (95% CI: –8.4, −4.2) and BDI-II score (n = 49) on average by −6.1 (95% CI: −8.2, −4.0; Figure 3). The impact of fatigue, measured by the mFIS (n = 51), showed a mean change of –8.2 (95% CI: −13.1, −3.3) from baseline, respectively (Figure 3). This decrease in mFIS score indicated a decrease in the impact of fatigue in terms of physical, cognitive and psychosocial functioning in RRMS patients with depression. Disability, as measured by the EDSS (n = 53), remained stable with a mean change of −0.1 (95% CI: −0.3, 0.1; screening mean ± SE, 3.5 ± 0.2; end of study mean ± SE, 3.4 ± 0.2; Figure 2). The TSQM questionnaire score remained at a similar level from baseline to end of study, change from baseline (n = 51) was 1.1 (95% CI: −0.8, 2.9).

PRIMUS (QoL score), PRIMUS (activities score) and EDSS change from baseline or screening to end of study.

BDI-II, HAM-D21 and mFIS change from baseline to end of study.
Over half of the patients (58%) were not employed. During the study period, eight patients were on sick leave due to MS and seven due to depression. A total of seven patients had MS-related changes in their employment status (reduced working hours, n = 4; reduced salary,n = 3) and six had depression-related changes in their employment status (reduced working hours, n = 3; reduced salary, n = 3). Moreover, 25% of the patients needed assistance by caregivers or from family and friends at the end of the study. A total of 10 patients required special equipment due to MS, such as walkers, wheelchairs and beds. Overall, one patient needed outpatient services due to depression, but none required hospitalisation, rehabilitation or nursing care.
At the end of the study, six patients reported to have experienced relapses in the past 3 months. However, 58% of the patients did not visit a physician outside the study and 73% did not need further medication for MS. In 75% patients no doctor’s appointment was required due to depression, and 89% did not need further medication.
Discussion
This study evaluated the safety and tolerability of the combination of fingolimod with three commonly prescribed antidepressants in patients with RRMS and comorbid depression. Even though only 54 patients could be enrolled, REGAIN is among one of the larger studies in patients with MS and comorbid depression. Unfortunately, as the planned sample size could not be recruited in the study, these findings should be interpreted with caution.
No unexpected safety signals were identified. Cardiac monitoring was recommended for 6 hours following the first dose of fingolimod, with extended monitoring in patients developing significant bradycardia and in those on medications known to prolong the QT interval [Dimarco et al. 2014]. SSRIs and SNRIs are also known to have potential cardiovascular side effects [Pacher and Kecskemeti, 2004]. In a meta-analysis conducted by Beach and colleagues, a mild and significant increase in QTc intervals during SSRI therapy compared with placebo (6.1 ms; 95% CI, 3.47–8.73) was described, especially for citalopram, although absolute prolongations (>450 ms) were only rarely seen [Beach et al. 2014]. Venlafaxine, through its action on catecholamine neurotransmission, can lead to increased blood pressure, sinus tachycardia and prolonged QTc interval, and may require cardiac monitoring [Stahl et al. 2005]. Both fluoxetine and citalopram have been shown to be associated with bradycardia [Pacher and Kecskemeti, 2004]. According to the European Medicines Agency in 2011, citalopram may be associated with QT prolongation [Pharmacovigilance-Working-Party, 2011]. Notably, with the limited data available, our study did not reveal clinically significant ECG changes for the combination of fingolimod and antidepressants. Clinically meaningful differences between antidepressants could not be observed (Table 4), possibly due to small patient numbers at respective timepoints. A recent post hoc analysis of safety data from the pooled fingolimod phase II and core phase III (FREEDOMS, FREEDOMS II and TRANSFORMS) studies assessed cardiac outcomes during treatment initiation in patients who were or were not receiving an SSRI [Bermel et al. 2015]. Consistent with our observation, coadministration of SSRIs and fingolimod was not associated with an increased incidence of abnormal ECG findings compared with fingolimod therapy alone [Bermel et al. 2015]. The START (STudy to vAlidate telemetRic ECG systems for firsT dose administration of Fingolimod) study conducted in Germany, which evaluated the first dose cardiovascular safety of fingolimod during 1 week in RRMS patients, had more than 10% patients who received concomitant SSRI. An interim analysis confirmed the good cardiac safety profile of fingolimod, including the patients who were treated with SSRIs [Limmroth et al. 2015].
The efficacy parameters QoL, depression and fatigue showed improvement and patients reported to be satisfied with the treatment, with a similar score at baseline and at the end of the study. Disability, as measured by EDSS, remained stable. However, the study was not powered to detect a difference in efficacy outcomes, and due to the small number of patients and short treatment duration, the data must be interpreted with caution.
Another limitation of our study is the lack of a control group receiving fingolimod treatment alone. Since it has been shown that fingolimod can improve depression in MS [Fox et al. 2014], we cannot rule out that beneficial effects attributed to coadministration with antidepressants in our study are, at least in part, caused by fingolimod.
Few published studies have evaluated the safety and efficacy of antidepressants in patients with MS and depression [Schiffer and Wineman, 1990; Ehde et al. 2008; Solaro et al. 2013]. In a double-blind study, 28 patients with MS and major depressive disorders were randomized to receive either a 5-week treatment of desipramine plus individual psychotherapy (n = 14) or placebo plus psychotherapy (n = 14). Compared with placebo, desipramine significantly reduced the HAM-D21 scores; however, side effects limited desipramine dosage in half of the treated patients [Schiffer and Wineman, 1990]. In another randomized double-blind study, higher reductions in the HAM-D21 score were observed with paroxetine treatment than with placebo; however, the treatment difference was not significant [Ehde et al. 2008]. In an open-label study in 75 patients with MS and depression, duloxetine (an SNRI) significantly reduced BDI and mFIS scores after 4 and 12 weeks of treatment [Solaro et al. 2013]. A few other smaller, open-label studies have also reported beneficial effects of antidepressants in patients with MS [Scott et al. 1995; Benedetti et al. 2004]. Consistent with these observations, in the current study coadministration of antidepressants with fingolimod had a beneficial effect on depression-related parameters such as QoL and fatigue in MS patients with comorbid mild-to-moderate depression. The improvement in depression, QoL and fatigue impact may improve an overall outlook of MS patients with depression; however, the study was not powered to detect a statistical difference in the efficacy parameters. Antidepressants may also play a role in reducing inflammatory activity and relapses in MS patients by reducing stress. There have been two randomized, placebo-controlled studies (n = 48 and n = 40, respectively) that demonstrated that SSRIs can be beneficial in MS patients without major depression [Mostert et al. 2008; Mitsonis et al. 2010]. Patients receiving antidepressants had fewer relapses [Mitsonis et al. 2010] and decreased magnetic resonance imaging (MRI) lesions [Mostert et al. 2008] compared with those receiving placebo. However, further evaluation using a well-designed study with a larger sample size is required to confirm these findings.
The REGAIN study did not aim to propose one particular SSRI or SNRI antidepressant over another as this study aimed to mirror a practical setting, evaluating safety and tolerability of SSRI/SNRI antidepressant therapy of fingolimod-treated RRMS patients. However, a preselection of commonly used antidepressant drugs was made, since increasing the number of antidepressants may have given us very imbalanced treatment arms.
Overall, the study was limited by the lower than planned number of enrolled patients and the results must be interpreted with caution. This low sample size may be partially due to the two DHCP Letters published at the time of the study conduct regarding cardiac safety monitoring of fingolimod and the cardiac side effects of citalopram (QT prolongation). These monitoring requirements may have reduced the readiness of investigators and the willingness of patients to participate, especially as no such monitoring was mandatory in routine clinical practice. Assignment of the antidepressant based on the investigator’s discretion is another limitation of this study, which may have resulted in selection bias.
Conclusion
In the present study, no new safety signals emerged for the combination of fingolimod with the most commonly used antidepressants (citalopram, fluoxetine and venlafaxine) in patients with RRMS and comorbid mild-to-moderate depression. Fatigue and QoL improved, and disability scores remained stable, although the results must be interpreted with caution because of the small number of patients and the rather short treatment duration.
As safety data for fingolimod and concomitant antidepressants (SSRIs or SNRIs) in patients with MS and depression are scarce, despite the data being limited, our study can add to the body of knowledge.
Footnotes
Appendix
In addition to the authors, the REGAIN Study Group comprises the following investigative teams: Joachim Koppai-Reiner, MD, Praxis Hunnenburgweg, 10 35510 Butzbach, Germany; Erik Strauss, MD, Neuroscience Center Leipzig, Fichtestraße 9, 04275 Leipzig, Germany; Beatrix Haerting, MD, Gemeinschaftspraxis, Willy-Brandt-Platz 4, 46045 Oberhausen, Germany; Stephan Wiehler, MD, Gemeinschaftspraxis, Obernstr. 5, 33602 Bielefeld, Germany; Johannes Boehringer, MD, Praxis für Neurologie und Psychiatrie, Hauptstr. 102, 33647 Bielefeld, Germany; Peter Laumen, MD, Praxis für Neurologie und Psychiatrie, An der Waad 6, 52499 Baeseweiler, Germany; Angelika Christopher, MD, Praxis Myslowitzer Str. 59, 12621 Berlin, Germany; Uwe Pfeiffer, Pfalzklinikum Klinik für Neurologie, Weinstraße 100, 76889 Klingenmünster, Germany; Werner Hofmann, MD, Praxis Elisenstr. 32, 63739 Aschaffenburg, Germany; Rolf Horn, MD, Praxis Alexander-von-Humboldt-Str. 30, 53604 Bad Honnef, Germany; Thorsten Lauter, MD, Studiengesellschaft Lauter - Spieker – Orbasli Hans-Böckler-Str. 23-27, 44787 Bochum, Germany; Udo Polzer, MD, Asklepios Fachklinikum Stadtroda Abteilung für Neurologie, Bahnhofstr. 1a, 07646 Stadtroda, Germany; Frank Halbgewachs, MD, Praxis Clichystr. 6, 89518 Heidenheim a. d. Brenz, Germany; Arno Siever, MD, Praxis Stau 1, 26122 Oldenburg, Germany; Holger Honig, MD, Klinikum Bremerhaven Reinkenheide, Postbrookstr. 103, 27574 Bremerhaven, Germany; Andreas Mahler, MD, Praxis Goethestr. 3, 28832 Achim, Germany; Karsten Kuhlgert, Medizinisches Versorgungs-zentrum Mittelweser Kliniken GmbH, Ziegelkampstr. 39, 31582 Nienburg, Germany; Martin Weber, Praxis Schillerstr. 3, 79576 Weil am Rhein, Germany; Felix Bischof, MD, Universitätsklinikum Tübingen, Neurologische Klinik und Hertie-Institut für klinische Hirnforschung, Hoppe-Seyler-Straße, 372076 Tübingen, Germany; Olaf Martin Hoffmann, MD, St. Josefs-Krankenhaus Potsdam, Klinik für Neurologie, Allee nach Sanssouci 7, 14471 Potsdam, Germany; Stefan Merkelbach, MD, Heinrich-Braun-Klinikum Zwickau GmbH, Klinik für Neurologie, Karl-Keil-Str. 35, 08060 Zwickau, Germany; Kirn Ralf Kessler, MD, NeuroCentrum am Kreiskrankenhaus, Am Ziegelkamp 1f, 41515 Grevenbroich, Germany; Roland Wenzelburger, MD, Zentrum für Neurologische Studien ZNS, Kiel Dänischenhagener Str. 12b, 24161 Altenholz, Germany; Geert Mayer, MD, Hephata Klinik Schimmelpfengstr. 2, 34613 Schwalmstadt-Treysa, Germany; Matthias Strittmatter, MD, SHG Klinik Merzig Abteilung Neurologie, Trierer Str. 148, 66663 Merzig, Germany.
Acknowledgements
Contributing members of the REGAIN Study Group are listed in the Appendix. The authors were assisted in the preparation of the manuscript by Dr. Hemant Kumar Mittal, and Sivaram Vedantam, Novartis Healthcare Private Limited, Hyderabad, India. Writing support was funded by the study sponsor. This study is registered at ClinicalTrials.gov with clinical trial identifier number NCT01436643.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Novartis Pharmaceu-ticals, Germany.
Conflict of interest statement
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Antonios Bayas received honoraria for consulting and/or as a speaker from Merck Serono, Germany; Biogen, Germany, Poland; Bayer Vital, Germany; Novartis, Germany, Austria; Sanofi/Genzyme, Germany, Netherlands; Roche, Germany and TEVA, Germany, for trial activities from Merck Serono, Germany; Biogen, Germany and Novartis, Germany and grants for congress trips and participation from Novartis, Germany; Biogen, Germany; Sanofi/Genzyme, Germany; and Merck Serono, Germany. Katrin Schuh, Monika Baier and Stefan Viktor Vormfelde are employees of Novartis Pharma GmbH, Nuremberg, Germany.