
Abstract
Fingolimod, the first oral disease-modifying therapy (DMT) approved for the treatment of multiple sclerosis (MS) and the only sphingosine 1-phosphate receptor modulator approved for any disease state, represents an important addition to the expanding DMT options for patients with MS. In three large phase III clinical trials, fingolimod 0.5 mg reduced relapses by approximately half compared with either placebo or weekly intramuscular interferon β1a. The risks associated with the use of fingolimod include first-dose bradycardia, macular edema, and elevation of liver enzymes; fingolimod may increase the risk of infections, some serious in nature, and potentially cause fetal harm. Breakthrough disease or intolerance of injectable medications may be factors that influence the initiation of fingolimod. Identification of potential patients suitable for fingolimod treatment requires a thorough understanding of the potential risks and the particular fingolimod indication of the national authority. To minimize risk, recommended baseline assessments that should be made prior to fingolimod initiation include complete blood count, liver transaminase levels, total bilirubin levels, electrocardiogram (ECG), ophthalmologic examination, varicella zoster infection status, and for women, childbearing potential. First-dose observation is required for all patients for at least 6 h, with hourly pulse and blood pressure measurements and ECG before and 6 h after the first dose. In the European Union, continuous telemetry monitoring is recommended. Healthcare providers should be aware of the potential for symptomatic bradycardia and the need for continuous overnight ECG monitoring for those at higher risk for bradycardia. With experience in over 63,000 patients and over 73,000 patient-years of exposure in clinical trials and postmarketing use, the benefits and full safety profile of fingolimod continue to become better elucidated. This information will enable healthcare providers to initiate fingolimod in appropriately selected and screened patients with MS.
Introduction
Fingolimod is an important addition to the expanding disease-modifying therapy (DMT) options available to treat patients with relapsing multiple sclerosis (MS). Fingolimod is the first oral DMT approved for the treatment of MS [Chun and Brinkmann, 2011], which may enhance patient adherence over that observed with parenteral therapies. Its efficacy has been well established in three phase III clinical trials: the FTY720 Research Evaluating Effects of Daily Oral Therapy in Multiple Sclerosis (FREEDOMS) trial [Kappos et al. 2010], the FREEDOMS II trial [Calabresi et al. 2012], and the Trial Assessing Injectable Interferon versus FTY720 Oral in Relapsing-Remitting Multiple Sclerosis (TRANSFORMS) [Cohen et al. 2010], each of which enrolled over 1000 patients with MS. Although both the 0.5-mg and 1.25-mg doses of fingolimod were examined in these clinical trials, the lower dose was found to be equally efficacious to the higher dose, with an improved safety profile. The annualized relapse rate (defined as the number of confirmed relapses per year) was reduced by 54% with fingolimod 0.5 mg versus placebo in FREEDOMS [rate 0.18, 95% confidence interval (CI) 0.15–0.22 with fingolimod 0.5 mg and 0.40, 95% CI 0.34–0.47 with placebo] [Kappos et al. 2010] and by 48% with fingolimod 0.5 mg versus placebo in FREEDOMS II [Calabresi et al. 2012]. The FREEDOMS trial also demonstrated a 30% reduction in the probability of disability progression with fingolimod versus placebo, as confirmed after 3 months [Kappos et al. 2010]. In TRANSFORMS, the annualized relapse rate was reduced by 52% with oral fingolimod 0.5 mg versus intramuscularly injected interferon β1a 30 µg (rate 0.16, 95% CI 0.12–0.21 with fingolimod 0.5 mg and 0.33, 95% CI 0.26–0.42 with interferon β1a), with a trend, but not a statistically significant difference, in confirmed disability progression between the treatments at 1 year [Cohen et al. 2010].
Fingolimod is the only sphingosine 1-phosphate (S1P) receptor modulator approved for any disease state. S1P receptors have five identified subtypes: S1P1-3 are found on cells of the immune, cardiovascular, and central nervous systems; S1P4 is found on lymphoid and hematopoietic tissues; and S1P5 is found predominantly on oligodendrocytes. T and B lymphocytes contain high levels of S1P1 expression. Functions of S1P receptors include lymphocyte recirculation, neurogenesis, neural cell migration, natural killer cell migration, endothelial cell function, vasoregulation, and cardiovascular development [Chun and Hartung, 2010; Gasperini and Ruggieri, 2012]. Exposure to fingolimod induces downregulation of S1P receptors within hours, including those on the surface of lymphocytes. The efficacy of fingolimod is likely due to sequestration of primarily naïve and memory T lymphocytes within lymph nodes, which reduces lymphocyte presence in the blood [Chun and Hartung, 2010]. In fact, temporary, reversible lymphopenia is a common consequence of fingolimod treatment [Gasperini and Ruggieri, 2012]. Fingolimod readily crosses the blood-brain barrier because it is highly lipophillic [Foster et al. 2007]. Mounting evidence suggests a potential central nervous system role of fingolimod, as S1P receptors are present on oligodendrocytes, astrocytes, and neurons [Chun and Hartung, 2010; Gasperini and Ruggieri, 2012].
Patient selection
In considering fingolimod therapy, healthcare providers must understand that patient selection is very important. Regulatory authorities from different countries provide varying guidelines for potential candidates for therapy. In the United States, for example, fingolimod is approved rather broadly for ‘patients with relapsing forms of MS’ (Gilenya; Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA). Therefore, the US Food and Drug Administration (FDA)–approved indication permits first-line use of fingolimod. In contrast, in the European Union, fingolimod is indicated for patients with highly active relapsing- remitting MS that fall into two categories. The first category is patients who have failed to respond to a full and adequate course (normally at least one year of treatment) of beta-interferon. Patients should have had at least 1 relapse in the previous year while on therapy, and have at least 9 T2-hyperintense lesions on cranial magnetic resonance imaging (MRI) or at least 1 gadolinium-enhancing lesion. A ‘non-responder’ could also be defined as a patient with an unchanged or increased relapse rate or ongoing severe relapses, as compared to the previous year.
The other category is patients with rapidly evolving severe relapsing remitting multiple sclerosis defined by 2 or more disabling relapses in one year, and with 1 or more gadolinium-enhancing lesions on brain MRI or a significant increase in T2 lesion load as compared to a previous recent MRI.
These categories are specified by the European Medicines Agency (EMA; http://www.ema.europa.eu/ema). In Canada, prescribing information states that Gilenya is ‘generally recommended in MS patients who have had an inadequate response to, or are unable to tolerate, one or more therapies for multiple sclerosis’ [Novartis, 2012].
Disease course factors play a role in the clinical decision to select fingolimod as a DMT. If a patient has experienced breakthrough relapses on another DMT, it is appropriate to consider alternative treatment, as a significant reduction in relapse rates may be achieved with a change in therapy [Rio et al. 2012]. In the Rebif versus Glatiramer Acetate in Relapsing MS Disease (REGARD) trial, which compared subcutaneous interferon β1a with subcutaneous glatiramer acetate [Mikol et al. 2008], and the Betaseron Efficacy Yielding Outcomes of a New Dose (BEYOND) trial, which compared subcutaneous interferon β1b with subcutaneous glatiramer acetate [O’Connor et al. 2009], only a minority of patients developed a relapse over the course of 2 years on either type of interferon or on glatiramer acetate (38% in REGARD; 40–42% in BEYOND). Therefore, two relapses within the same year, or annual relapses, is a subpar clinical response. As noted above, the annualized relapse rate in the TRANSFORMS trial, which enrolled patients with a history of at least one relapse during the previous year or at least two relapses during the previous 2 years, was 52% lower with oral fingolimod 0.5 mg than with intramuscular interferon β1a 30 µg over the course of 1 year. Furthermore, fingolimod should be considered for patients with worsening disability and relapses on a current DMT. New MRI activity with or without new clinical symptoms might also prompt a switch in treatment to fingolimod, depending on the patient’s country of residence. Mounting data support the increased risk of worsening disability with the development of new T2 and enhancing lesions on MRI while on treatment [Prosperini et al. 2009; Rio et al. 2008, 2009].
Many factors regarding patient dissatisfaction with current treatment may influence a decision to initiate fingolimod. Patients who are highly needle phobic have opted for this first oral MS treatment. Many of these patients with needle phobia have struggled with adherence to injectable medications, which resulted in periods of treatment disruption or inconsistent administration [Turner et al. 2009]. Some patients have switched to fingolimod due to ongoing injection site reactions, including reactions with more severe complications, such as injection site necrosis or lipoatrophy. Uncontrolled side effects of injectable DMTs, such as flu-like symptoms, depression, and immediate post-injection reactions, may also be a consideration. Patients receiving natalizumab who are positive for John Cunningham virus (JCV) antibody may consider a switch to fingolimod to reduce the risk of progressive multifocal leukoencephalopathy (PML), a potentially fatal viral brain infection. The JCV antibody–positive patients at greatest risk for PML have received greater than 2 years of natalizumab treatment and have had previous exposure to immunosuppressive agents [Bloomgren et al. 2012].
Conversely, some factors may dissuade the use of fingolimod. Patients at high risk for bradycardia or heart block may meet fingolimod contraindication criteria or fall into categories specified in treatment guidelines as requiring increased monitoring for first-dose observation (see below). Hypertension is a potential risk of fingolimod therapy [Novartis, 2012]. Patients with inadequately controlled hypertension should seek medical treatment before initiating fingolimod. Collaboration with the patient’s primary care provider in the selection of the antihypertensive agent(s) is useful in order to avoid the use of β blockers and heart rate–lowering, calcium-channel blockers when initiating fingolimod. There is no limitation on administering these agents in patients already treated with fingolimod.
People with diabetes are likely at higher risk for the development of macular edema with fingolimod. In earlier renal transplant studies that used fingolimod doses 5- to 10-fold higher than the dose approved for MS, patients with diabetes had a higher rate of macular edema (30%) compared with patients without diabetes (4%) [Jain and Bhatti, 2012]. Patients with MS and diabetes mellitus were excluded from the phase III clinical trials, so the true incidence of macular edema in patients with diabetes who are receiving fingolimod 0.5 mg is unknown. While diabetes is not a contraindication to fingolimod therapy, patients with diabetes, especially those with baseline visual impairment from retinopathy or previous optic neuritis, should be aware of this increased risk. Implementation of a schedule of more routine ophthalmologic evaluations once such patients have begun fingolimod therapy would be prudent. Patients with uveitis are also at increased risk for macular edema [Novartis, 2012].
Fingolimod is classified by the FDA as pregnancy category C. Women of childbearing potential should be informed of the potential risk of fetal harm on fingolimod therapy. Only limited data exist on the use of fingolimod in pregnant women. Data from rats have shown teratogenicity, most commonly, persistent truncus arteriosis and ventricular septal defect after fingolimod administration to pregnant animals. Risk of pregnancy should be minimized with an effective method of birth control [Novartis, 2012].
S1P receptors exist on airway smooth muscle cells. Potential respiratory effects on patients with severe reactive airway disease are not well defined since such patients were excluded from the fingolimod clinical trials. The effect of fingolimod on long-term tumor surveillance has not been established.
Clinicians also need to be aware of the still-developing safety profile of fingolimod, especially with its first-in-class mechanism of action. Postmarketing case reports include tumefactive lesions [Visser et al. 2012] on fingolimod therapy, worsening of MS with fingolimod therapy given after natalizumab [Centonze et al. 2012], varicella zoster encephalitis with fingolimod treatment [Ratchford et al. 2012], and MS rebound after varicella zoster infection and discontinuation of fingolimod [Gross et al. 2012]. However, no causal relationship between these cases and fingolimod could be established, and these cases must be placed in the context of the growing postmarketing experience of greater than 63,000 patients exposed. The long-term efficacy of fingolimod in clinical practice also remains to be established, and clinical effectiveness trials are needed. Ongoing extensions of the phase II and phase III clinical trials and new fingolimod trials will help clarify further the full safety and efficacy profile.
Baseline safety assessments
To maximize safety with the use of fingolimod, key assessments should be performed before first-dose observation (Table 1). Baseline complete blood count should be obtained within 6 months of fingolimod initiation [Novartis, 2012]. Patients with leukopenia at baseline could be at a potential increased risk of serious infection once they initiate fingolimod. If a patient has a low white blood cell count while on current therapy such as interferon, consider holding treatment for 2 weeks prior to the first dose of fingolimod and repeating the white blood cell count just prior to first-dose observation. Analysis of the relationship between lymphocyte count and infection incidence has shown that infection of any type did not appear to be related to lymphocyte count [Francis et al. 2010]. No specific requirements for monitoring white blood cell counts on therapy have been established. Absolute lymphocyte counts during therapy fall generally into a range of 0.2–0.8 × 109/liter. The measurement of lymphocyte subsets, such as CD4 cells, during therapy has not been validated as a useful practice, as counts would be expected to be low, based on the mechanism of action of fingolimod [Novartis, 2012].
Baseline assessments required prior to initiation of fingolimod.
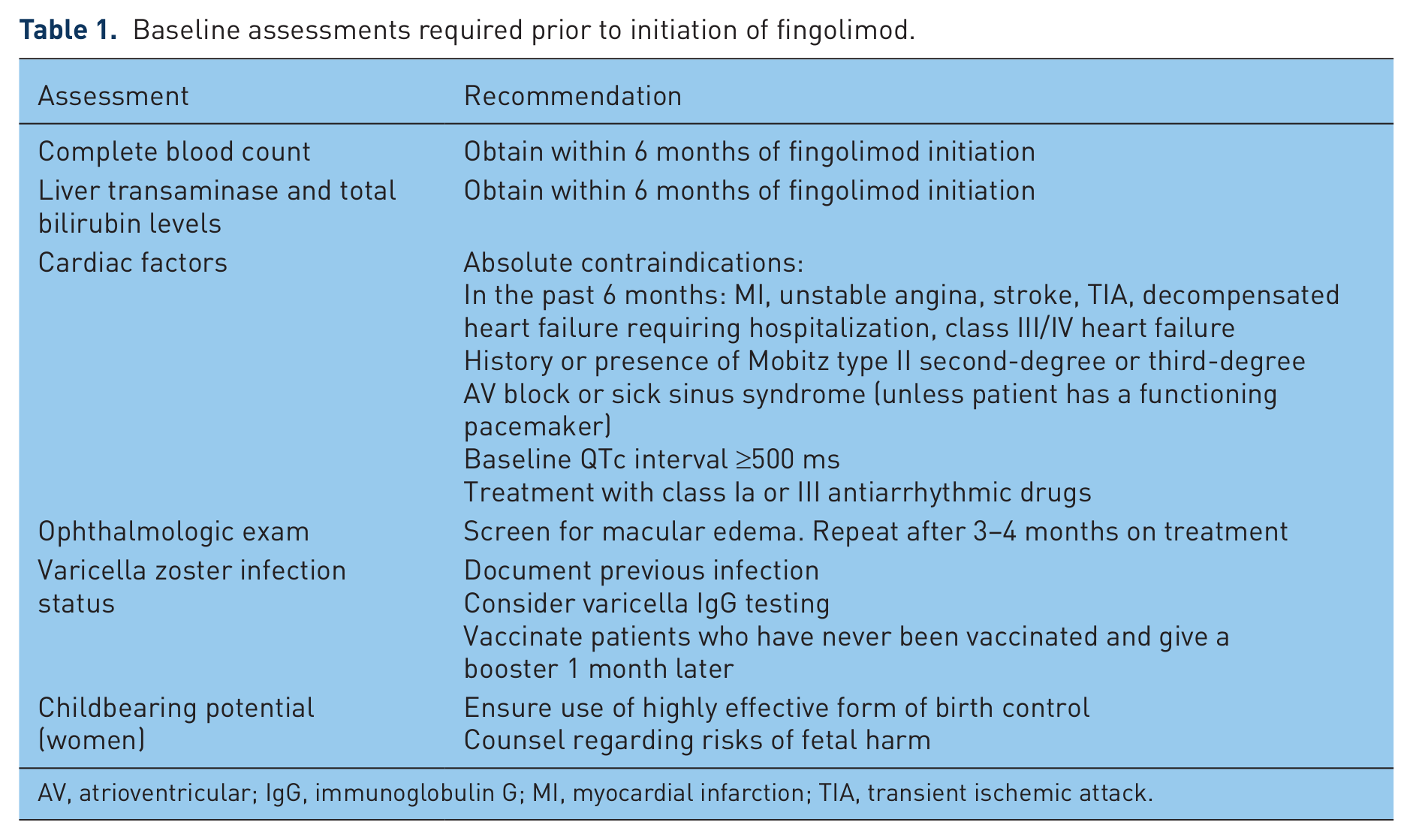
AV, atrioventricular; IgG, immunoglobulin G; MI, myocardial infarction; TIA, transient ischemic attack.
Liver transaminases (aspartate transaminase and alanine transaminase) plus total bilirubin levels should be obtained within 6 months of fingolimod initiation [Novartis, 2012]. If levels were high on a previous treatment, allowing levels to normalize prior to initiating fingolimod would be prudent. Due to the potential risk of elevated liver enzymes, the risks of fingolimod therapy may likely outweigh the benefits if significant baseline liver disease is known.
A critical component to fingolimod initiation is screening for cardiac factors that may place patients at risk for complications on fingolimod during first-dose administration. Absolute contraindications for fingolimod include recent (within the last 6 months) myocardial infarction, unstable angina, stroke, transient ischemic attack, decompensated heart failure requiring hospitalization, and/or class III/IV heart failure. Cardiac contraindications for fingolimod use also include a history or presence of Mobitz type II second- degree or third-degree atrioventricular (AV) block, or sick sinus syndrome (unless the patient has a functioning pacemaker). In addition, fingolimod is contraindicated in patients with baseline QTc interval of at least 500 ms or patients being treated with class Ia or III antiarrhythmic drugs [Novartis, 2012].
An ophthalmologic evaluation should be performed at baseline and after 3–4 months on fingolimod to assess for macular edema. Patients found to have uveitis on the screening exam would be at increased risk for macular edema if fingolimod is initiated. If macular edema is found during fingolimod therapy, generally fingolimod discontinuation would be recommended.
Varicella status is another important baseline assessment. In the phase III clinical trial, a death from disseminated primary varicella zoster virus infection occurred in a woman who had no previous exposure to varicella and had never been vaccinated against varicella. Confounding factors were administration of the 1.25-mg fingolimod dose (instead of the approved 0.5-mg dose) and treatment with intravenous and oral steroids [Novartis, 2012]. Documentation of previous varicella exposure is important to prevent the risk of a primary infection. Serology testing for varicella immunoglobulin G titers can provide further confirmation of status, since occasionally a patient with a history of exposure does not have adequate titer to test positive on the commercially available enzyme-linked immunosorbent assay [Katial et al. 1999]. Since varicella vaccination utilizes a live-attenuated virus, vaccination and subsequent booster 1 month later should occur at least 1 month prior to initiation of the first dose of fingolimod [Novartis, 2012].
A woman’s childbearing potential should be addressed and the method of preventing pregnancy documented. An effective form of birth control should be used with consultation with the patient’s gynecologist if needed. The patient needs to be counseled about the risks of fetal harm and informed that fingolimod must be discontinued for 2 months before trying to conceive [Novartis, 2012].
First-dose observation
First-dose monitoring is required for all patients. Patients should be observed for at least 6 h with hourly pulse and blood pressure measurements. The monitoring facility must have adequate resources to manage symptomatic bradycardia [Novartis, 2012]. Current settings for monitoring have included outpatient office settings and hospital infusion centers. New EMA and FDA guidelines require an ECG prior to dosing and at 6 h after the first dose. In addition, continuous telemetry monitoring is recommended in the European Union (http://www.ema.europa.eu/ema).
Extended monitoring is required for certain patients. If the heart rate is at its lowest recorded level at 6 h, further monitoring is required until the heart rate begins to increase from the nadir. Extended monitoring to resolution should be implemented with new-onset second-degree or high AV block at 6 h or a heart rate less than 45 beats per minute at 6 h. If symptomatic bradycardia arises, start appropriate management and begin continuous ECG monitoring until resolution. For those patients requiring pharmacologic treatment, admission to a medical facility is required for overnight continuous ECG monitoring. If a second dose of fingolimod is given to a patient with symptomatic bradycardia that required pharmacologic intervention, repeat first-dose procedures need to be followed [Novartis, 2012].
Continuous overnight ECG monitoring is required for certain patients at higher risk for bradycardia who do not have absolute contraindications for fingolimod therapy (as described above). Such patients include individuals with a history of ischemic heart disease, myocardial infarction, cerebrovascular disease, symptomatic bradycardia, recurrent syncope, severe untreated sleep apnea, and AV and sinoatrial heart block. To determine whether these patients are appropriate for first-dose observation, cardiology consultation should be obtained. Continuous overnight monitoring is to be implemented for those patients with an increased risk for prolonged QT interval. Patients with prolonged baseline QTc interval (>450 and <500 ms for men, >470 and <500 ms for women), at risk for QT prolongation (hypokalemia, hypomagnesemia), or on concomitant QT-prolonging drugs with a known risk of torsades de pointes should also be monitored overnight with continuous ECG. Examples of medications with a risk of torsades de pointes are citalopram, chlorpromazine, haloperidol, methadone, and erythromycin. A continuous overnight monitoring protocol should be implemented for individuals receiving concurrent therapy with drugs that slow heart rate or AV conduction (e.g. β blockers, heart rate–lowering calcium-channel blockers such as diltiazem or verapamil, or digoxin) [Novartis, 2012].
Transient effects on heart rate are explained by transient agonism of S1P receptors in myocytes [Sanna et al. 2004]. Across clinical trials (fingolimod 0.5 mg, n = 1,640; phase II, FREEDOMS, FREEDOMS II, and TRANSFORMS and their extensions), the incidence of symptomatic bradycardia was 0.5% [Novartis, 2012], and bradycardia responded to administration of atropine or isoprenaline when necessary. The incidence of second-degree AV block was 0.1%, and most cases were asymptomatic [Novartis, 2012]. Pooled phase III clinical trial data for fingolimod 0.5 mg documented that only 1.4% of patients had their lowest pulse rate in the 40–44 beat per minute range (0.3% for patients on placebo), and no patients had a recorded pulse rate below 39 beats per minute [DiMarco et al. 2012]. Pooled phase III 6 h postdose 12-lead ECG data demonstrated an incidence of 4.7% for first-degree AV block, 0.2% for Mobitz type I second-degree AV block, and no cases of 2:1 second-degree block with fingolimod 0.5 mg [DiMarco et al. 2012].
Summary and conclusion
Fingolimod is a very effective treatment for relapsing forms of MS, with proven efficacy across three large phase III clinical trials. The FREEDOMS trial demonstrated a 54% reduction in relapses and a 30% reduction in the likelihood of disability progression. As the first oral DMT for MS, fingolimod provides a more convenient and palatable route of administration for patients. Patient selection requires a thorough understanding of the potential risks of this S1P-receptor modulator. Fingolimod should not be administered to MS patients with absolute contraindications. Appropriate monitoring should be implemented for higher-risk individuals. With mounting postmarketing experience in over 63,000 patients, the benefits and full safety profile of fingolimod continue to become better elucidated. Armed with knowledge, healthcare providers have the opportunity to initiate fingolimod in appropriately selected and screened patients.
Footnotes
Acknowledgements
Editorial assistance for this manuscript was provided by Complete Healthcare Communications, Inc. (Chadds Ford, PA, USA).
Funding
Editorial assistance for this manuscript was funded by Novartis Pharmaceuticals Corporation.
Conflict of interest statement
Barry A. Singer, MD, was an investigator in the FREEDOMS II and TRANSFORMS studies. He has received honoraria from Novartis Pharmaceuticals Corporation for speaking and consulting.