
Abstract
Laquinimod is a novel immunomodulatory agent, in development as a potential disease-modifying treatment for multiple sclerosis (MS). Structurally related to linomide, its pharmacological predecessor, laquinimod combines anti-inflammatory and possibly clinically relevant neuroprotective effects with the convenience of oral administration. In this review we aim to highlight the immunomodulatory and neuroprotective effects of laquinimod, and to describe its effects in animal models of MS. Furthermore, we focus on current results of clinical studies in MS. Randomized, controlled clinical trials in relapsing MS demonstrate a dose–response effect on disease activity, measured by reduced clinical relapse rate, reduced number of brain MRI active lesions, as well as on sustained disability and brain atrophy. Laquinimod has a favourable tolerability and safety profile. A new phase III study, recently completed, will soon provide further details on the therapeutic potential of this drug. Laquinimod is a promising emerging treatment for relapsing–remitting MS that may provide a new therapeutic option in the near future.
Background
Multiple sclerosis (MS) is a chronic immune-mediated disease affecting the central nervous system (CNS), and characterized by demyelination and axonal damage. The estimated prevalence of MS is approximately 2.5 million cases worldwide and the disease is the second most common cause of neurological disability in young adults [Patwardhan et al. 2005]. Most people with MS are diagnosed between the ages of 20 and 40. MS initially begins with a relapsing–remitting course characterized by unpredictable acute attacks, called exacerbations, with worsening of symptoms followed by full, partial, or no recovery of function, but later evolves into progressive disease. If untreated, MS typically leads to substantial accumulation of both physical and cognitive disability over time. For that reason, early use of disease-modifying therapies (DMTs) is required. DMTs for MS should reduce relapse rate, limit the onset of new demyelinating lesions and postpone the development of long-term disability. Until recently all available therapies, albeit with excellent safety, were moderately effective and were injectable. Parenteral application of these drugs is associated with bothersome side effects and low patient compliance. Thus, there remains an unmet need for the development of more effective and well-tolerated oral therapies for the treatment of MS. Based on results of large randomized, controlled double-blind trials, a number of oral medications have already been or soon will be submitted for marketing authorization in several countries, and have already been approved in some countries. As of today, these include four immunomodulators: fingolimod (Gilenya©), teriflunomide (Aubagio©), dimethyl fumarate (Tecfidera©) and laquinimod (LAQ). The development of these drugs with improved efficacy, better tolerability and oral administration has received a new impetus with the widening of new therapeutic perspectives in MS.
Mechanism of action of laquinimod
LAQ (5-chloro-N-ethyl-4-hydroxy-1-methyl-2-oxo-N-phenyl-1, 2-dihydroquinoline-3-carboxamide), also known as ABR-215062, is structurally related to linomide (roquinimex), which demonstrated efficacy in murine acute experimental autoimmune encephalomyelitis (EAE) and in MS [Diab et al. 1998; Zhu et al. 1998; Karussis et al. 1996]. Owing to severe immune-mediated side effects (serositis, pericarditis, myocardial infarction) observed during phase III trials [Noseworthy et al. 2000; Wolinsky et al. 2000], the clinical development of linomide was halted. Among several roquinimex-related 3-quinolinecarboxamide derivatives, tested in murine EAE and for induction of pro-inflammatory reaction in the beagle dog, LAQ showed improved potency and superior toxicological profile compared with the lead compound linomide [Jönsson et al. 2004], with no apparent propensity to induce inflammatory reactions. LAQ was shown to dose-dependently inhibit the development of both acute and chronic EAE models [Brunmark et al. 2002; Yang et al. 2004; Aharoni et al. 2012; Ruffini et al. 2013; Jolivel et al. 2013] and by direct comparison based on dose and total exposure, LAQ was approximately 20 times more potent than the immunomodulator linomide [Brunmark et al. 2002; Yang et al. 2004].
The exact mechanism of action of LAQ is not fully elucidated, although based on animal models LAQ has been suggested to reduce leukocyte migration into the CNS by downregulation of VLA-4-mediated adhesiveness, inhibiting Th17-proinflammatory responses and also by modulating the cytokine balance in favour of Th2/Th3 cytokines interleukin (IL)-4, IL-10 and transforming growth factor (TGF)-β [Wegner et al. 2010; Yang et al. 2004; Zou et al. 2002; Aharoni et al. 2012; Jolivel et al. 2013]. In vitro experiments on human B cells from healthy or MS patients have shown that LAQ modulates B-cell activity, probably by promoting an IL10+ CD86+ CD25+ B-cell subset with regulatory features, along with other immunomodulatory mechanisms such as modulation of B-cell cytokine production and B-cell-mediated T-cell cytokine profile [Toubi et al. 2012]. Furthermore, in LAQ-treated mice Foxp3+ T-regulatory cells were 2.2- to 2.5-fold augmented within the overall inflammatory cells and the T cells in the CNS lesions [Aharoni et al. 2012], respectively, in comparison with untreated mice.
Recently it has been suggested that the beneficial effect of LAQ may be mediated by modulation of dendritic cells (DCs) affecting the antigen presentation capacity [Gurevich et al. 2010; Thöne et al. 2012; Jolivel et al. 2013]. Under treatment with LAQ, both in murine models and in patients with MS, a reduction in the number of DCs has been observed, in addition to a reduced ability of these cells to induce the proliferation of CD4 + T cells as well as to stimulate the secretion of pro-inflammatory cytokines [Jolivel et al. 2013]. Treatment with LAQ of mature DCs results in reduced production of chemokines with a consequent impact on the migratory properties of monocytes [Jolivel et al. 2013]. In vivo LAQ treatment modulates subpopulations of myeloid antigen-presenting cells (APC), including a decrease in DCs and an elevation of anti-inflammatory type II monocytes [Schulze-Topphoff et al. 2012]. Recently, in a cuprizone-induced demyelination model with wild type and Rag-1-deficient mice, LAQ treatment was associated with a dose-dependent reduction of callosal demyelination, microglial density, axonal damage, reactive gliosis and oligodendroglial apoptosis. These effects were interpreted as a purely central mechanism of LAQ, mediated by downregulation of astrocyte pro-inflammatory response. Several experimental studies converge on the hypothesis that the biological effects of the LAQ are mainly mediated via inhibition of NF-κB pathway [Gurevich et al. 2010; Brück et al. 2012; Jolivel et al. 2013].
Evidence is also accumulating that LAQ might have a neuroprotective effect in EAE and in MS, which is not only due to the effects of immunomodulation, but also to a direct effect of LAQ in the CNS [Aharoni et al. 2012; Ruffini et al. 2013]. LAQ treatment induces in situ overexpression of brain-derived neurotrophic factor (BDNF) in the cortex and basal ganglia of chronic EAE mice, as compared with untreated mice. This is associated with significantly reduced myelin and axonal damage and with preservation of the CNS tissue [Aharoni et al. 2012]. Axonal protection by LAQ was also observed in a rat model of optic neuritis [Sühs, 2007]. Also in patients with MS, LAQ treatment results in a significant and persistent increase in BDNF serum levels when compared with baseline and placebo-treated patients [Thöne et al. 2012]. Furthermore, it was demonstrated that the attenuation of EAE by LAQ to be mediated by the reduction of overactive NMDA receptors of presynaptic terminal, while increasing GABAergic synaptic currents. This dual effect may limit glutamatergic excitotoxicity, and the axonal loss is consequently limited in EAE mice compared with controls [Ruffini et al. 2013].
Efficacy data of laquinimod in multiple sclerosis
Following the promising results of the preclinical studies, several randomized, controlled trials (RCTs) have been performed to further establish the efficacy of LAQ in MS (Table 1).
Efficacy and safety/tolerability data of laquinimod in multiple sclerosis clinical trials.
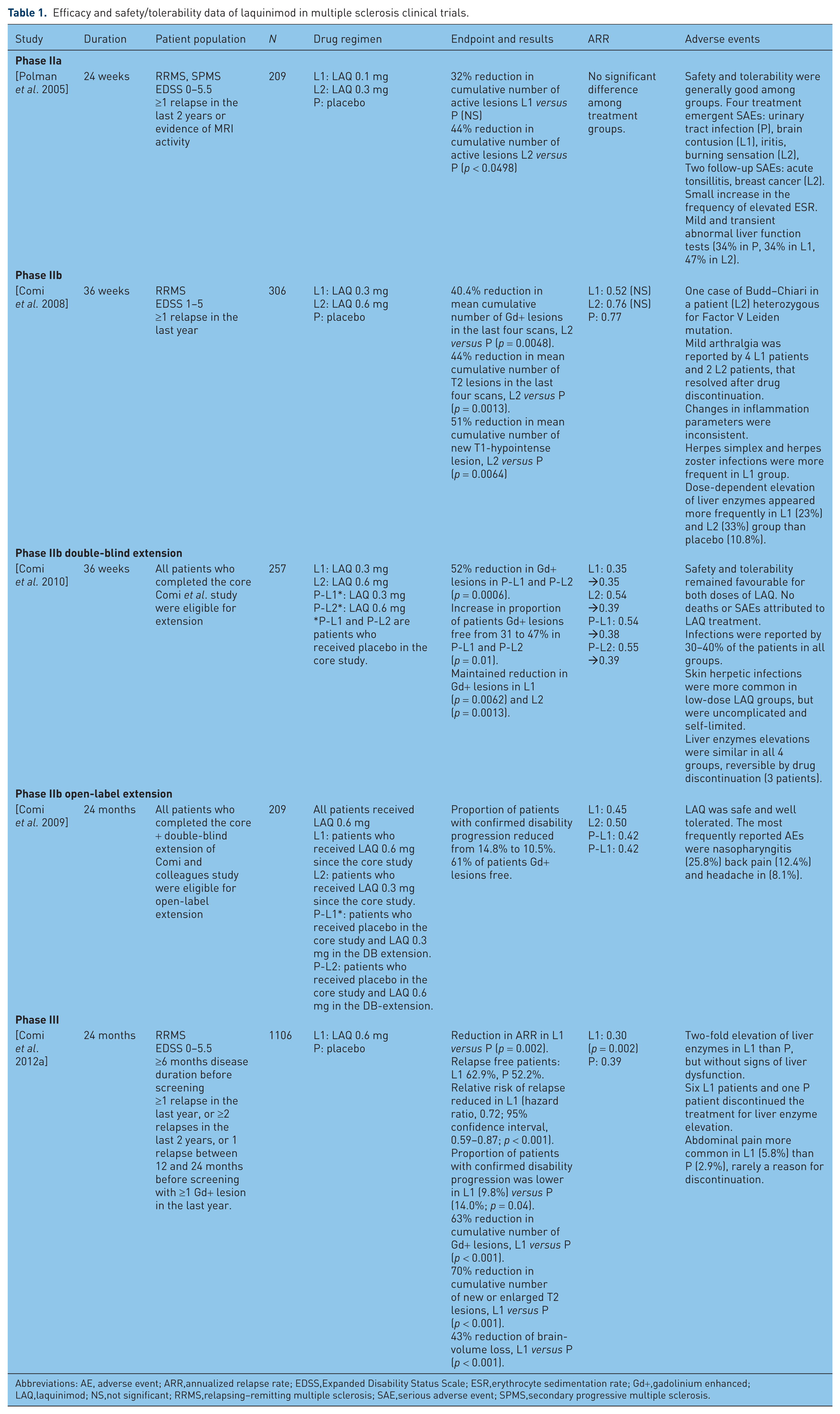
Abbreviations: AE, adverse event; ARR,annualized relapse rate; EDSS,Expanded Disability Status Scale; ESR,erythrocyte sedimentation rate; Gd+,gadolinium enhanced; LAQ,laquinimod; NS,not significant; RRMS,relapsing–remitting multiple sclerosis; SAE,serious adverse event; SPMS,secondary progressive multiple sclerosis.
Two phase I studies were conducted to obtain safety and pharmacokinetics data, and the dosages of 0.1 and 0.3 mg/day were selected for subsequent clinical trials [Polman et al. 2005]. The first published report was a 24-week, multicentre, double-blind, placebo-controlled trial in patients with relapsing–remitting MS (RRMS) or secondary progressive MS (SPMS) [Polman et al. 2005]. Patients were required to have Expanded Disability Status Scale (EDSS) scores between 0 and 5.5, evidence of disease activity based on MRI, and/or a clinical relapse in the 2 years prior to enrolment. Of 256 screened patients, 209 were randomized to receive LAQ 0.1 mg, LAQ 0.3 mg or placebo, as three daily tablets for 24 weeks. The primary endpoint efficacy variable was the cumulative number of MRI active lesions (new enhancing lesions and new or enlarging T2 lesions) over 24 weeks. Safety measures included adverse events, physical examination and laboratory variables. Treatment with LAQ 0.3 mg resulted in significant suppression (44%; p = 0.0498) of MRI activity compared with placebo as for per-protocol analysis, whilst the intention-to-treat analysis did not show significance. In the subgroup of patients with at least one gadolinium-enhanced (Gd+) lesion at baseline, the reduction of new lesions was larger (52%; p = 0.005). No significant difference in the primary endpoint was achieved between LAQ 0.1 mg/day and placebo. No significant group differences with respect to clinical variables (relapses, disability) were found; on the other hand the study was not powered to assess these parameters.
The phase IIb, multinational, multicentre, randomized, double-blind, parallel-group, placebo-controlled trial included 306 ambulatory patients, with EDSS score between 1 and 5, with at least one relapse in the last year and with at least one Gd+ lesion on brain MRI scans [Comi et al. 2008]. In view of LAQ dose-related effects observed in previous preclinical and clinical observations, higher dosages were chosen, LAQ 0.3 and 0.6 mg, and compared with placebo over 36 weeks. The primary efficacy outcome measure was the cumulative number of Gd+ lesions on the last four triple-dose Gd-MRI scans of the treatment period (weeks 24, 28, 32 and 36). Secondary endpoints hierarchically included the following: (a) cumulative number of Gd+ lesions on week 12, 16, 20, 24, 28, 32 and 36 scans; (b) number of T2 lesions on week 24, 38, 32 and 36 scans; (c) the total number of confirmed relapses during the double-blind treatment period. The LAQ 0.6 mg arm showed a significant reduction by 40.4% of Gd+ lesions per scan in the last four scans as compared with placebo (p = 0.0048). Patients in the LAQ 0.6 mg dose arm also had a 44% reduction in the cumulative number of new T2 lesions in the last four scans compared with placebo (p = 0.0013), as well as a 51% reduction of new T1-hypointense lesions (p = 0.0064). With regard to clinical parameters, patients who received LAQ 0.6 mg showed an annual relapse rate (ARR) of 0.52 ± 0.92 versus 0.77 ± 1.25 in the placebo group (p = 0.0978). The LAQ 0.3 mg showed no significant results versus placebo neither for primary nor for any secondary endpoints. The disappointing results achieved in the LAQ 0.3 mg group were judged as ‘surprising’ by the investigators, and the explanations given were of a greater sensitivity of the triple-dose Gd-MRI used in the previous trial compared with the single dose of this trial, in addition to a possible slower onset of anti-inflammatory action of the low dose as compared with the high dose [Comi et al. 2008]. Upon completion of the 36th week of the study, 257 (91%) patients entered a double-blinded extension phase, and patients originally in the placebo arm were randomized to receive either LAQ 0.3 or 0.6 mg, while the other patients already in the LAQ-treated arms continued at the same dose [Comi et al. 2010]. Patients underwent clinical assessments at weeks 4, 12 and every 12 weeks thereafter, and brain MRI was performed at weeks 0 and 36. The main outcome measures were the number of Gd+ lesions and the number of new hypointense T1 lesions on enhanced scans (black holes). By the end of the extension phase, patients switched from placebo to either LAQ 0.3 or 0.6 mg dose gained a mean reduction of 52% (p = 0.0006) of Gd+ lesions as compared with week 0, and the rate of these patients with Gd+ free scans increased from 31% to 47% (p = 0.01). Also for the patients who continued either on LAQ 0.3 or 0.6 mg the mean number of Gd+ lesions was reduced significantly (p = 0.0062; p = 0.0013), with a trend of larger effect for the high dose, while the rate of inactive scans was unchanged for both doses from the end of the placebo-controlled phase to the end of the extension phase. The effects on new T2 lesions and new T1-hypointense lesions, as well as T2 lesion volume was larger for the higher LAQ dose, but did not reach significance. Likewise, the decrease in ARR observed in the initial LAQ 0.6 mg arm appeared sustained throughout the extension phase, while in the other groups no significant reduction was observed. A further 24-month open-label extension to the 18-month double-blind core plus extension of the placebo-controlled study was conducted [Comi et al. 2009]. All 209 patients entering the open-label extension were treated with LAQ 0.6 mg, and a total of 155 patients have thus completed 42 months of study follow up. During the 24-month extension, 10.5% of patients met confirmed disability progression on EDSS compared with 14.8% during the first 18 months. The MRI-measured disease activity remained low with 61% of patients free of Gd-enhancing lesions at month 42.
The clinical development of LAQ continued in two multinational, multi-centre, 2-year, placebo-controlled phase III clinical trials, designed to evaluate the safety, efficacy and tolerability of LAQ 0.6 mg once daily in patients with RRMS [Comi et al. 2012a; Vollmer et al. 2011]. The primary outcome of both studies was to assess efficacy, as measured by number of confirmed relapses. In the first phase III trial (the ALLEGRO study), 1105 patients were randomized (1:1) to receive LAQ or placebo [Comi et al. 2012a]. Treatment with LAQ as compared with placebo was associated with a modest reduction in the mean (±SE) ARR (0.30 ± 0.02 versus 0.39 ± 0.03; p = 0.002) and with a reduction in the risk of confirmed disability progression (11.1% versus 15.7%; hazard ratio [HR] 0.64; 95% confidence interval [CI] 0.45–0.91; p = 0.01). The mean cumulative numbers of Gd+ lesions and new or enlarging T2 lesions were lower for patients receiving LAQ than for those receiving placebo (1.33 ± 0.14 versus 2.12 ± 0.22 and 5.03 ± 0.08 versus 7.14 ± 0.07, respectively; p < 0.001 for both comparisons). As an exploratory endpoint, patients receiving LAQ showed a lower percentage of brain-volume loss from baseline to 24 months, as compared with the placebo group (adjusted mean, –0.87% versus –1.30%; mean difference, 0.43 percentage points; p < 0.001) [Comi et al. 2012a]. The 2-year double-blind phase of ALLEGRO study was followed by a 36-months open-label extension phase in which all patients (n = 424 early start, n = 417 delayed start) received LAQ 0.6 mg [Comi et al. 2012b]. The ARR of the early start patients was 0.21 in the extension phase, compared with the 0.30 ARR observed during the placebo-controlled phase of the ALLEGRO study. After 12 months of LAQ treatment, the ARR of the delayed start patients was 0.24 compared with 0.39 of the placebo arm [Comi et al. 2012b]. Overall, during the 36 months of the ALLEGRO study, risk of confirmed disability progression was significantly reduced for early start patients compared to delayed start patients (11.8% versus 16.7%, HR = 0.62, p < 0.0038). Likewise during the 36 months of the study, there was a lower proportion of patients with disability progression for the early start compared with delayed start patients for the subgroup that has completed 1 year in the OL phase (14.5% versus 21.2%, p = 0.0123) [Comi et al. 2013]. The second phase III trial (the BRAVO study) of LAQ 0.6 mg in RRMS, compared LAQ with placebo in a double-blind design and to intramuscular interferon-β-1a (30 µg once weekly) in a rater-blinded design [ClinicalTrials.gov identifier: NCT00605215] in 1331 patients with RRMS (1:1:1). At 24 months, the ARR did not reach statistical significance, but showed a trend toward reduction on LAQ (risk ratio [RR]=0.823, 95% CI 0.664–1.020, p = 0.075). However, because of significant dissimilarities between treatment groups at baseline in T2 lesion volume and the rate of patients with Gd+ lesions, adjusted analyses were carried out. LAQ was thus shown to significantly reduce ARR (0.37 and 0.29 for placebo and LAQ, respectively, RR=0.787, 95% CI 0.637–0.972, p = 0.026), as well as the risk of disability progression as measured by the EDSS (HR=0.665, 95% CI 0.447–0.989, p = 0.044) and brain atrophy on MRI (27.5%, p < 0.0001). LAQ was shown to modestly reduce the mean cumulative numbers of new or enlarging T2 lesions compared with placebo (–19%, p = 0.037), while a marked reduction in confirmed 24-week EDSS progression was observed in the LAQ group over placebo (–40.6%, p = 0.04). For interferon-β-1a IM, following a similar correction, ARR was reduced (0.27) compared with placebo (29%, p = 0.002), as well as the mean cumulative number of Gd+ lesions (–60%, p < 0.0001) and new or enlarging T2 lesions (–52%, p < 0.0001). No treatment effect of interferon-β-1a IM was observed on brain atrophy and disability progression was reduced (28.7%, p = 0.089) [Vollmer et al. 2011]. Full results from the trial are expected to be published shortly. Both phase III studies are continuing in open-label extensions.
Safety and tolerability profile of laquinimod in multiple sclerosis
LAQ has generally been very well tolerated in the trials published to date (Table 1), and none of the LAQ-treated patients experienced any of the SAEs (serositis, cardiovascular events and thrombosis) observed during linomide clinical trials, or any life-threatening malignancies and opportunistic infections [Comi et al. 2012a].
In the phase IIa trial LAQ treatment was safe and well tolerated at both doses of 0.1 and 0.3 mg, with a rate of SAEs comparable with placebo [Polman et al. 2005]. There were no clinically relevant changes in inflammatory parameters. A mild and transient increase of abnormal liver function tests was reported (34% in placebo and LAQ 0.1 mg groups, 47% in LAQ 0.3 mg group).
In the phase IIb trial only two SAEs were judged potentially as attributable to LAQ, one case (LAQ 0.6 mg) of Budd–Chiari syndrome in a patient heterozygous for the Factor V Leiden mutation and one case (LAQ 0.3 mg) with marked elevation of liver enzymes, but with no clinical signs of liver failure [Comi et al. 2008]. The elevations of liver enzymes were dose-dependent and reversible after treatment discontinuation. In the 36-week extension of this trial safety and tolerability remained favourable for both doses of LAQ. There were no deaths or SAEs attributed to LAQ treatment. Arthralgia occurred more frequently in LAQ 0.3 mg treatment group (5.0%), as only one case reported as severe. Infections were reported by 30–40% of the patients in all groups and skin herpetic infections showed similar incidence to the core study, however they were uncomplicated and self-limited. Liver enzymes elevations were comparable in all four groups, reversible by drug discontinuation and without signs of liver failure. In the open-label extension of this trial [Comi et al. 2009] the safety and tolerability profiles continued to be excellent. Nasopharyngitis, back pain and headache were all more commonly reported in LAQ-treated patients, although these were not considered SAEs.
In the phase III ALLEGRO trial, liver enzymes elevations, typically seen within the first 6 months of therapy, occurred twice as frequently in the LAQ group [Comi et al. 2012a]. These elevations were transient and not accompanied by liver failure signs. Elevated levels of alanine aminotransferase led to discontinuation in patients receiving LAQ and in patients receiving placebo. No deaths and no other SAEs were observed. Safety data from the open-label extension of the ALLEGRO study, from the BRAVO study core phase and its open-label extension study are not yet available.
Discussion
There is a high demand for innovative and at the same time orally available MS therapeutics. LAQ is a promising emerging treatment for RRMS, combining anti-inflammatory and possibly clinically relevant neuroprotective effects with the convenience of oral administration. LAQ is immunomodulatory, without suppressive effects on the immune system. Recently, new biological effects have been attributed to LAQ, in particular in the peripheral regulation of antigen presentation capacity and in the central modulation of astrocyte inflammatory activity. Clinical studies have confirmed, in addition to a statistically significant efficacy on ARR and new MRI lesion load, although not always confirmed, interesting beneficial effects on sustained disability and MRI measures of brain-tissue loss. LAQ has shown an excellent tolerability and a favourable side effect profile. Furthermore, no special requirement is needed for monitoring, which is a clear advantage over comparators.
Nevertheless, some issues deserve caution in assessing the effectiveness of LAQ and in framing the possible role it may play in the future treatment scenario of MS. LAQ has a peculiar mechanism of action compared with other drugs, and this may confer unique protective effects. Indeed, the effect of LAQ effect on disability progression and brain atrophy appear more pronounced than its effect on relapses and new lesion formation. It is not clear yet how to interpret this apparent dichotomy, and whether these interesting results on disability are interpretable as independent by the effect of LAQ on inflammatory activity. Furthermore, the applicability of the trial results to the general MS population is limited, due to the narrow patient population included in the studies that also did not include patients previously receiving immunosuppressive treatments. Furthermore, still a relatively small body of evidence to support long-term safety is available. Just as was the case with other treatments in MS, other low-frequency side effects may become apparent after LAQ is approved in large post-marketing trials.
The decision to use a novel oral drug such as LAQ will most likely be based on an overall assessment of efficacy, safety, tolerability and adherence, the potential need for monitoring and cost effectiveness. Although better patient compliance is expected compared with the injectable drugs, the safety profiles will have to be watched carefully. The BRAVO trial and the two phase III extension trials hopefully will provide further information about its efficacy, also compared with an actual first-line treatment (interferon-β-1a) in MS.
On August 2012, the manufacturers announced that, following a written agreement reached with the US Food and Drug Administration on a Special Protocol Assessment, a third phase III study of LAQ in RRMS patients will be initiated. The third phase III LAQ trial CONCERTO will evaluate two doses of the investigational product (0.6 and 1.2 mg) in approximately 1800 patients for up to 24 months. The primary outcome measure will be confirmed disability progression as measured by the EDSS. The CONCERTO study is ongoing since the first quarter of 2013. Moreover, in July 2012 the company submitted a marketing authorization application for LAQ in RRMS to the European Medicines Agency. If the application is successful, LAQ could be approved in Europe by year-end 2013 (see http://www.activebiotech.com/laquinimod-new-promising-treatment-of-multiple-sclerosis#sthash.biEHSi5.dpuf).
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
Claudio Gasperini received speaking honoraria from Biogen Idec, Novartis, Teva, Merck-Serono.