
Abstract
The therapeutic landscape for multiple sclerosis (MS) is rapidly changing. Currently, there are eight FDA approved disease modifying therapies for MS including: IFN-β-1a (Avonex, Rebif), IFN-β-1b (Betaseron, Extavia), glatiramer acetate (Copaxone), mitoxantrone (Novantrone), natalizumab (Tysabri), and fingolimod (Gilenya). This review will highlight the experience to date and key clinical trials of the newest FDA approved agents, natalizumab and fingolimod. It will also review available efficacy and safety data on several promising therapies under active investigation including four monoclonal antibody therapies: alemtuzumab, daclizumab, ocrelizumab and ofatumumab and three oral agents: BG12, laquinimod, and teriflunomide. To conclude, we will discuss where each of these new therapies may best fit into treatment algorithms.
Keywords
Introduction
The therapeutic landscape for multiple sclerosis (MS) is rapidly changing. The first disease modifying therapy, interferon-β-1b (IFN-β-1b) was introduced in 1993. Currently, there are eight FDA approved disease modifying therapies for MS including: IFN-β-1a (Avonex, Rebif), IFN-β-1b (Betaseron, Extavia), glatiramer acetate ((GA) Copaxone), mitoxantrone (Novantrone), natalizumab (Tysabri) and fingolimod (Gilenya). There has been significant global experience with the first-line injectable therapies (IFN-β-1a, IFN-β-1b and GA) and their side effect profiles have been well characterized. The relative safety and efficacy of these therapies has provided prescribers with a level of comfort in recommending them to patients with MS. Injectable therapies can be uncomfortable and inconvenient for patients. The new and emerging agents provide alternative modes of administration including oral agents and intravenous infusions along with improved efficacy; however, these therapies are not without risks.
The newest FDA approved agents that have been added to the MS treatment armamentarium are natalizumab and fingolimod. Natalizumab was initially FDA approved in 2004, as a once monthly intravenous infusion with multiple clinical trials demonstrating efficacy in the treatment of relapsing forms of MS. It was withdrawn from the market in 2005 after it was found to be associated with progressive multifocal leukoencephalopathy (PML) a rare, opportunistic brain infection. It was subsequently reintroduced to the market in 2006 with a black box warning for PML. 1 The neurology community learned an important lesson from the experience with natalizumab in that newer disease modifying therapies are not without serious adverse events, some of which can be life-threatening. In 2010, fingolimod became the first FDA approved oral agent for treatment of relapsing remitting MS (RRMS). Clinical trials demonstrated that fingolimod is efficacious in reducing the frequency of MS relapses and in decreasing MRI markers of disease activity; however, there were increased risks of infection including two deaths from herpes virus infections in the original phase III trial with one case of herpes virus encephalitis and one case of primary disseminated varicella zoster. 2 Appropriate screening prior to drug initiation and close monitoring may reduce these risks.
Increased efficacy in decreasing relapse rates and MRI disease markers of activity are exciting to clinicians and patients, however, caution will need to be used when selecting the appropriate therapy for each patient. A careful balance of benefits versus risks will need to be considered with each individual patient's disease profile. This review will highlight the experience to date and key clinical trials of the newest FDA approved agents, natalizumab and fingolimod, along with promising therapies under active investigation for MS including four monoclonal antibody therapies (Tables 1 and 2): alemtuzumab, daclizumab, ocrelizumab and ofatumumab and three oral agents (Tables 3 and 4): BG12, laquinimod, and teriflunomide.
Summary of New and Emerging Monoclonal Therapies.

Optimal dose under investigation,
Efficacy of New and Emerging Monoclonal Therapies.

Summary of New and Emerging Oral Therapies.

Optimal dose under investigation.
Efficacy of New and Emerging Oral Therapies.

Monoclonal Antibodies
Alemtuzumab
Alemtuzumab is a humanized monoclonal antibody which targets CD52, an epitope expressed on T and B lymphocytes, natural killer cells and most monocytes, but it is not expressed on hematopoietic precursors.3,4 Treatment with alemtuzumab results in rapid depletion of CD52 containing cells by antibody dependent cellular toxicity. 5 Following treatment, reconstitution of these cell populations is staggered with return to baseline for monocytes and B cells at 3 months, CD8+ T cells around 30 months and CD4+ T cells at approximately 61 months. 6
Clinical studies
Early investigations by Coles et al of alemtuzumab treatment in secondary progressive MS (SPMS) indicated that it may be useful in relapse suppression rather than prevention of disability progression.
7
This finding led to the development of CAMMS223, a randomized, single-blind, double-dummy phase 2 clinical trial of patients with early RRMS which compared the efficacy and safety of annual intravenous cycles of alemtuzumab at a dose of 12 mg or 24 mg versus subcutaneous IFN-β-1a at a dose of 44 μg three times weekly. The co-primary outcome measures were time to sustained accumulation of disability (SAD) measured by the Expanded Disability Status Scale (EDSS) maintained over 6 months and annualized relapse rate (ARR). The data was analyzed based on pooled data for both treatment doses of alemtuzumab. Six month SAD was significantly reduced in the alemtuzumab arm versus IFN-β-1a (9% vs. 26.2%;
The successful phase 2 program led to the development of the parallel phase 3 clinical trials. The first phase 3 trial, CARE-MS I, was a 2-year randomized, rater-blinded, double-dummy, active comparator clinical trial studying the efficacy and safety of annual alemtuzumab (12 mg intravenously administered daily for 5 days during the first year and for 3 days in year 2) to subcutaneous IFN-β-1a 44 mcg 3 times weekly over the course of 2 years in treatment naïve patients with RRMS. Co-primary outcomes were ARR and time to 6-month SAD. The first primary endpoint was met with a 55% reduction in ARR with alemtuzumab treatment compared to treatment with IFN-β-1a (
The second phase 3 trial coined, CARE-MS II, utilized the same design as CARE-MS I, however, in this trial enrollment criteria required that patients have had at least 2 relapses within 2 years prior to entering the trial with at least 1 relapse within the year prior to enrollment and 1 relapse while on a MS disease-modifying therapy. Co-primary endpoints were the same as CARE-MS I, and this trial met both of its primary endpoints with a 49% relapse rate reduction compared to IFN-β-1a treated patients (
Safety
Patients treated with alemtuzumab in CAMMS and CARE MS trials experienced significantly more infusion related reactions and drug-induced autoimmunity compared to those treated with IFN-β-1a. Infusion associated reactions include fever, headache, malaise and/or urticarial rash, fortunately, these symptoms are largely prevented by pre-treatment with corticosteroids. 12 Drug-induced autoimmunity with alemtuzumab, most commonly involves thyroid dysfunction and occurs in 20%–30%.4,5 Additionally, there have been a few cases of Goodpasture's disease. 5 The most serious auto-immune condition which has occurred with alemtuzumab is idiopathic thrombocytopenic purpura (ITP). The index patient in the CAMMS-223 died secondary to ITP when early signs were not reported. A monitoring risk management program is now in place for all patients treated with alemtuzumab to identify early signs of ITP. 4
Daclizumab
Daclizumab is a humanized monoclonal antibody against the α subunit, CD25, of the IL-2 receptor on T cells, B cells, macrophages and natural killer cells. 13 Interleukin-2 plays a key role in T cell activation and proliferation. Cluster of differentiation-25 (CD-25) blockade selectively inhibits activated T cells which play an important role in the pathogenesis of auto-immune disease and therefore, this drug is of interest in the treatment of MS. In addition, daclizumab has been shown to increase the quantity of CD56bright natural killer (NK) cells (a regulatory subset of NK cells) which down regulate adaptive T cell responses. 14 Administration of 1 mg/kg of daclizumab every 4 weeks results in blockade of 95% of CD25 on T cells.
Clinical studies
The CHOICE study was a 6-month, randomized, double-blind, placebo-controlled, phase 2 clinical trial studying the efficacy of adding daclizumab to interferon beta in RRMS patients with active disease despite interferon monotherapy. Patients remained on interferon and were randomized to 1 of 3 treatment arms: add-on daclizumab 2 mg/kg subcutaneous every 2 weeks, add-on daclizumab 1 mg/kg subcutaneous every 4 weeks or add-on placebo. Patients were subsequently followed for an additional 48 weeks once the 6-month treatment period was completed. The primary endpoint was total number of new or enlarged gadolinium enhancing lesions on brain MRI measured every 4 weeks between weeks 8 and 24. There was a significant reduction in gadolinium enhancing lesions in the high dose daclizumab add-on treatment arm compared to add-on placebo (placebo 4.75 Gd + lesions vs. high dose-daclizumab 1.32 Gd+ lesions
The SELECT trial, a 1-year, randomized, double-blind, placebo-controlled, dose-finding phase 2b trial studying the efficacy and safety of daclizumab at 150 mg, 300 mg or placebo administered subcutaneously every 4 weeks in RRMS patients were recently released. The primary endpoint, ARR, was significantly lower in both high and low dose treatment arms compared to placebo (0.21, 0.23 vs. 0.46;
The DECIDE study is a 2-year, randomized, double-blind, parallel-group, active-control, phase 3 clinical trial in patients with RRMS exploring the efficacy and safety of daclizumab 150 mg subcutaneous injection once every 4 weeks compared to interferon β-1a intramuscular injection once weekly in RRMS patients which is currently enrolling.
A recent NIH study by Borges et al demonstrated a decrease in the rate of brain atrophy in patients with multiple sclerosis on daclizumab compared to placebo by assessing brain volume on T1-weighted and T2-FLAIR MRI sequences using TOADS-CRUISE program. Annual brain volume decreased by 3.1 cc compared to 5.1 cc per year in MS patients not on daclizumab.
17
Specifically, decreases in atrophy were primarily noted in the thalamus with a 38% reduction (
Safety
According to results from phase 2 clinical trials, daclizumab was well tolerated in patients with MS. Daclizumab in combination with interferon β in the CHOICE trial did not raise any additional safety concerns.15,18 In CHOICE, incidence of adverse events was similar in all treatment groups including infections (urinary tract infection, upper respiratory infection, nasopharyngitis, and sinusitis) which occurred in 68%–69% of all patients in the trial. Rash occurred in 18% in the add-on low dose daclizumab group, 8% in the add-on high dose daclizumab group and 8% in the add-on placebo group. No opportunistic infections or deaths occurred during the study period. Two patients in the daclizumab treatment groups developed malignancy. One patient with a family history of breast cancer developed ductal breast cancer in situ 1 year after the last dose of daclizumab and another developed recurrence of a prior condition, pseudomyxoma peritonei. 15 Cutaneous events occurred in 24% of patients in the daclizumab arm and in 6% in the placebo arm.
In SELECT, adverse events were similar in the treatment arms compared to placebo with the main adverse events including infection 2% vs. 0%, cutaneous events 1% vs. 0% and liver function abnormalities greater than five times the upper limit of normal 4% vs. < 1% respectively. In SELECT, 1 patient death occurred secondary to a complication from a psoas muscle abscess in a patient who suffered a serious skin adverse event and there was also 1 death in an ongoing dose extension study, SELECTION, due to possible autoimmune hepatitis in the treatment arm. 19
Ocrelizumab
Ocrelizumab is a humanized, recombinant monoclonal antibody against CD20 on B cells. 20 It has been shown to enhance antibody dependent cell mediated cytotoxicity and leads to a reduction in complement dependent cytotoxicity similar to rituximab.21,22
Clinical studies
Kappos et al reported on a 24-week, randomized, double-blind, placebo-controlled, phase 2 clinical trial examining the efficacy and safety of ocrelizumab at 2 separate doses, 300 mg (low dose) and 1000 mg (high dose) intravenous, administered on day 1 and 15 compared to IFN-β-1a intramuscularly once weekly or placebo in RRMS patients. The study primary endpoint was total number of gadolinium enhancing lesions on T1 weighted MRI at weeks 12, 16, 20 and 24. An intention-to-treat analysis revealed a statistically significant decrease in total gadolinium enhancing lesions compared to placebo (low dose: 89% decrease (
As part of a dose-finding exploration, patients in the low dose ocrelizumab arm were switched to 600 mg at week 24 on day 1 and placebo on day 15, and at weeks 48 and 72 they received 600 mg on day 1 and there was no scheduled treatment on day 15 (Table 5). The high dose ocrelizumab arm was administered ocrelizumab 1000 mg at week 24 on day 1 and placebo on day 15, followed by 1000 mg on day 1 at weeks 48 and 72 (Table 5). Patients originally randomized to placebo or IFN-β-1a treatment arms were reassigned to receive ocrelizumab 300 mg on day 1 and 15 at week 24, followed by 600 mg on day 1 at weeks 48 and 72 (Table 1). Recently, Kappos et al presented the full 96 week follow up data from the study. The original low dose and high dose ocrelizumab treatment arms showed statistically significant reductions (
Ocrelizumab phase II trial design.

Safety
In the phase 2 trial, serious adverse events occurred in 2% in the 600 mg ocrelizumab group, 4% in the placebo group and 4% in the IFN-β-1a group. Serious infections occurred at similar rates in both ocrelizumab and placebo groups. Infusion related events occurred more frequently in the ocrelizumab treatment arms, 35% for the 600 mg group and 44% for the 2000 mg group, compared to placebo with 9% experiencing infusion reactions. Adverse events across all treatment groups were similar from weeks 24–48. No opportunistic infections occurred. One patient in the ocrelizumab 2000 mg treatment arm died at week 14, after developing systemic inflammatory response syndrome and requiring a prolonged hospital course. 23
Ofatumumab
Ofatumumab is a type I, humanized monoclonal (IgG1κ) antibody against a novel epitope of CD20 on B lymphocytes. It is believed to mediate B cell lysis by complement-dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity.24,25 It targets a CD20 epitope which is distinct from that targeted by rituximab, by binding both small and large extracellular loops of the CD20 surface antigen.25,26
Clinical studies
Sorensen et al conducted a 48-week, double-blind, placebo-controlled, phase 2 clinical trial investigating the safety and pharmacokinetics of ofatumumab in 38 RRMS patients randomized to receive 2 infusions of ofatumumab 100 mg, 300 mg, 700 mg or placebo at weeks 0 and 2. At 24 weeks, the placebo group was switched to 2 infusions of ofatumumab and the ofatumumab treatment groups were switched to receive placebo in a blinded fashion. Efficacy, the secondary endpoint, was assessed by MRI metrics. The combined ofatumumab group was found to have a mean cumulative number of new gadolinium enhancing lesions on monthly MRI of 0.04 between weeks 8–24 compared to 9.69 in the placebo group. The relative reduction in gadolinium enhancing lesions for the combined ofatumumab group was 99.8% compared to placebo (
Safety
In October 2009, the FDA gave ofatumumab fast track approval for the treatment of fludarabine and alemtuzumab resistant CLL. The most common adverse effects seen in the treatment of CLL include infusion reactions related to cytokine release syndrome. Other adverse effects reported are infection, neutropenia, anemia, rash, fever and diarrhea. 25 In the phase 2 trials in RRMS, no concerns for patient safety have been reported.27,29
Natalizumab
Natalizumab is a recombinant humanized monoclonal antibody which antagonizes the α4β1 integrin (also known as VLA-4) expressed on the surface of activated lymphocytes and monocytes. It is a selective adhesion molecule inhibitor which binds specifically to the α4 subunit. 1 This prevents activated leukocytes from adhering to and migrating across the endothelial blood brain barrier yielding its therapeutic effect in MS.
Clinical studies
In 2003, Miller et al conducted the first randomized, double-blind, placebo-controlled trial to evaluate the safety and efficacy of natalizumab in patients with RRMS and relapsing SPMS. Patients were randomized to treatment with natalizumab or placebo given every 28 days for 6 months.
30
The primary endpoint, number of new brain lesions on monthly gadolinium enhanced MRI, revealed a marked decrease in the mean number of new enhancing lesions over the 6 month treatment period in both treatment groups compared to placebo: 0.7 per patient in the low dose natalizumab group (
The AFFIRM study, conducted by Polman et al was a 2-year, randomized, double-blind, placebo-controlled, phase 3 clinical trial exploring the efficacy and safety of natalizumab in 942 patients with RRMS. The study primary endpoints were achieved with significant reductions in the risk of sustained progression of disability (sustained for 3 months) by 42% over 2 years in the natalizumab arm compared to placebo (HR 0.58; 95% confidence interval 0.43 to 0.77,
Fox et al conducted the RESTORE study, a 24-week, randomized, partially placebo-controlled trial to investigate the effect of natalizumab treatment interruption on MS disease activity through clinical and radiologic parameters. Patients were randomized 1:1:2 to continue natalizumab, switch to placebo or to open label alternative treatment with interferon β-1a, glatiramer acetate or methylprednisolone. Preliminary findings of the 24 week randomization were presented at the European Committee for Treatment and Research In Multiple Sclerosis (ECTRIMS) triennial meeting in October 2011. Rescue treatment with natalizumab and/or high dose corticosteroids were offered to patients experiencing clinical relapse or 1 new gadolinium enhancing lesion > 0.8 cm3 or ≥2 gadolinium enhancing lesions. 32 Rescue therapy with natalizumab was given to 30% of patients due to disease activity and 29% of patients met MRI rescue criteria. Relapse occurred in 5% of patients in the natalizumab arm and 16%–29% in the other arms. 32 This study detected a high rate of recurrence of MS disease activity following treatment interruption in concordance with other studies.33,34
Combination studies
Rudick et al conducted the SENTINEL study, a 2-year, randomized, double-blind, placebo-controlled, phase 3 clinical trial to evaluate the efficacy and safety of natalizumab and IFN-β-1a combination therapy in comparison to IFN-β-1a monotherapy in 1171 patients with RRMS. The study's 1 year primary endpoint, ARR, was significantly reduced with combination therapy compared to IFN-β-1a monotherapy at 0.34 and 0.75 respectively (
Goodman et al carried out the GLANCE study, a 6-month, randomized, double-blind, placebo-controlled phase 2 clinical trial which evaluated the safety and tolerability of natalizumab in combination with GA in patients with relapsing MS. A total of 110 patients were randomized to combination therapy with natalizumab and GA versus placebo and GA. The study primary endpoint, rate of development of new active lesions (new gadolinium enhancing lesions and nonenhancing new or enlarging T2 lesions) revealed a decreased mean rate of new active lesions at 0.03 with combination versus 0.11 for GA alone (
Safety
Natalizumab was initially FDA approved for the treatment of relapsing forms of MS in the United States in November 2004. In February 2005, Biogen Idec, voluntarily withdrew the drug from the market after 2 patients with MS in the SENTINEL trial 35 and 1 patient with Crohn's disease (also on azathioprine) developed PML from pre-marketing clinical studies. Natalizumab was re-approved in June 2006 in the US and Europe as monotherapy for treatment of relapsing forms of MS with a black box warning about PML.1,37–39 In the US, the TOUCH prescribing program was set in place to restrict prescription and administration of natalizumab to registered pharmacies, physicians and infusion centers. It also provides guidelines for close patient monitoring for PML and other adverse events. 40
PML is a rare brain disease caused by opportunistic, lytic infection of oligodendroglial cells with JC virus, a human polyomavirus. 41 It occurs in immunocompromised individuals and is most commonly seen in patients with HIV, malignancy and organ transplantation in association with immune suppression. Recently, the use of monoclonal antibodies for autoimmune disease has led to an increased incidence of PML. Natalizumab does not directly suppress immunity, but is believed to alter central nervous system (CNS) immune surveillance. 42 StÜve et al studied the effect of natalizumab on CNS immune surveillance. Peripheral blood and CSF samples were collected from patients with MS on natalizumab treatment for at least 6 months and compared to controls with RRMS who were never on natalizumab and patients with other neurological disease. Multiple sclerosis patients on natalizumab were found to have significantly fewer white blood cells, CD4+ T cells, CD8+ T cells, CD19+ B cells and CD138+ plasma cells in their CSF in comparison to MS patients never treated with natalizumab. 42 Following 6 month cessation of natalizumab in MS patients originally treated with the drug, these cell counts remained significantly lower than those of MS patients not on natalizumab treatment. 42 A follow up study demonstrated no significant difference between CSF cell counts at 14 months after drug discontinuation between MS patients previously treated with natalizumab and MS patients never treated with natalizumab. 43 Peripheral blood lymphocytes from MS patients treated with natalizumab remained within normal limits throughout the study. This study demonstrated the prolonged effects of natalizumab on CSF lymphocyte populations and it supports the hypothesis of impaired CNS immune surveillance with natalizumab treatment.
As of February 1, 2012, natalizumab associated PML has been confirmed in 207 patients and 41 of these patients have died (21%). 44 A total of 95,300 patients have been exposed to natalizumab. Currently, the overall risk of PML is estimated to be 2.11 per 1000 patients. 44 For patients with a history of prior immune suppressant drug use, this risk increases to 3–4 times the risk of someone without prior immune suppressant use. 44 A significant increase in natalizumab associated PML incidence occurs after patients have received 24 doses. The risk of PML for patients who have received 25–36 infusions is 1.97, approximately four times the risk for a patient who has received 13–24 infusions. 44 PML is diagnosed by MRI and testing for the presence of JCV DNA in the CSF by PCR. If testing is negative, but clinical suspicion remains, natalizumab should be withheld and the patient should be closely monitored. Repeat CSF JCV PCR testing and MRI are recommended. Once a diagnosis of PML is made, the majority of patients undergo plasma exchange and/or immunoadsorption to hasten removal of natalizumab. Rapid removal of the drug often leads to the immune reconstitution inflammatory syndrome (IRIS) which is characterized by progressive decline in neurologic functioning. 45 In attempt to prevent IRIS, patients are typically treated with corticosteroids when they begin treatment with plasma exchange although this needs to be systematically studied. 45
A serum anti-JCV antibody assay was developed which combines ELISA with immunoadsorption to assist in risk stratification for the development of PML in patients on natalizumab and considering natalizumab. This test is currently undergoing clinical evaluation in the STRATIFY 2 study. Anti-JCV antibodies are detected in about 54% of MS patients and the annual seroconversion rate is between 2%–3%. 46 It was FDA approved on January 20, 2012 and is now commercially available.
After natalizumab was reintroduced to the market, patients from AFFIRM, 31 SENTINEL 35 and GLANCE 36 were invited to enroll in STRATA (Safety of Tysabri Re-dosing and Treatment) an ongoing study to assess the safety and efficacy of drug re-exposure following treatment interruption. At present, 4 cases of PML have been detected in this study after 33, 34, 44 and 46 doses of natalizumab and 3 of these patients had prior exposure to immunosuppressive treatment. 47 Between 22–30 months prior to their diagnosis of PML, serum samples were collected from these individuals. The samples were subsequently analyzed for anti-JCV antibody and all 4 were found to be positive. 46 This study is ongoing.
Two cases of primary central nervous system lymphoma (PCNSL) have been reported in MS patients on natalizumab.48,49 The possibility of a causal association between natalizumab and PCNSL remains unclear. Both cases occurred in 40 year-old men and the incidence of PCNSL in this age group is rare. 48 Infusion and hypersensitivity reactions can occur with natalizumab treatment. Infusion reactions include: headache, dizziness and nausea. These symptoms can be managed with pre-treatment with loratadine or acetaminophen and with slowing the rate of infusion. Hypersensitivity reactions include anaphylaxis, urticarial, allergic dermatitis, hives, fever, rash, rigors, pruritis, nausea, flushing, hypotension, dyspnea or chest pain. It is generally recommended that patients who experience hypersensitivity reactions discontinue use of natalizumab. 50 The AFFIRM study revealed that persistent antibodies to natalizumab are associated with increased infusion reactions. 31
Natalizumab is pregnancy level C and is not recommended for treatment in pregnant women. A recent study of 35 women who accidentally became pregnant while on natalizumab reported that 29 women gave birth to 28 healthy children, one child was born with hexadactyly, 5 women suffered early miscarriage and 1 woman chose to have an elective abortion. 51
Oral Agents
Fingolimod
Fingolimod is the first oral agent FDA approved for use in RRMS. Its novel mechanism of action, efficacy, and oral route of administration make it an attractive alternative to presently used therapies. Unlike other drugs used in MS and other autoimmune diseases, fingolimod modulates the immune response, creating a relative leukopenia that is reversible with discontinuation of the drug. This, theoretically, spares the body's ability to fight infection. Fingolimod is a sphingosine-1-phosphate (S1P) receptor modulator. S1P is a phospholipid that is primarily produced in the plasma by erythrocytes, but is ubiquitous in the body. 52 One of the roles of S1P is chemoattraction and motility, particularly with regard to lymphocytes. The active form of fingolimod, binds non-specifically to 4 of the 5 S1P receptor types. The S1P1 receptor is highly expressed on unactivated lymphocytes. Binding of fingolimod to the S1P1 receptor causes abnormal phosphorylation, resulting in internalization and degradadation of the receptor. The decrease in S1P1 receptor expression on plasma lymphocytes results in their sequestration in the lymph nodes, thus preventing lymphocyte activation and subsequent transit to sites of inflammation.
Fingolimod likely has other effects that may be responsible for its efficacy. The S1P1 and S1P3 receptors are expressed on astrocytes. In vitro studies of active and chronic MS lesions show an increase in S1P1 and S1P3 expression. In the experimental autoimmune encephalitis (EAE) model, injections of S1P into mice resulted in astrogliosis. Thus, a decrease in the expression of S1P may be protective to astrocytes in the CNS. 52 In addition, treatment of cultured human astrocytes with fingolimod resulted in decreased production of pro-inflammatory cytokines.
Clinical studies
Two pivotal trials, TRANSFORMS and FREEDOMS, demonstrated efficacy of fingolimod in treating RRMS. TRANSFORMS was a 1-year, randomized, double blind, double-dummy phase 3 trial which compared the effect of 0.5 mg and 1.25 mg doses of fingolimod to the effect of weekly intramuscular IFN-β-1a on clinical and MRI measures in MS. 53 The primary outcome measure of the study was ARR. Secondary outcome measures included number of new or enlarged T2 lesions and 3-month sustained progression of disability.
Annualized relapse rate in both high dose (0.2, 95% CI, 0.16 to 0.26) and low dose (0.16, 95% CI, 0.12 to 0.21), fingolimod groups was lower as compared to the IFN-β-1a group (0.33, 95% CI, 0.26 to 0.42;
ARR in TRANSFORMS and FREEDOMS.

The FREEDOMS study was a 2-year, phase 3, randomized, double-blind, trial with two doses of fingolimod, 1.25 mg and 0.5 mg, compared to placebo.
54
Again, the primary outcome measure of the study was ARR. The main secondary outcome measure was time to sustained 3-month disability. Other secondary outcome measures included MRI markers of disease activity. Both high dose (ARR = 0.16, 95% CI, 0.13 to 0.19) and low dose (0.18, 95% CI, 0.15 to 0.22) groups of fingolimod showed lower ARR as compared to placebo (0.40, 95% CI, 0.34 to 0.47;
Safety
Common and serious adverse effects were not significantly different among high and low dose fingolimod groups and either IFN-β-1a or placebo in TRANSFORMS or FREEDOMS.53,54 A dose-dependent decrease in heart rate of less than or equal to 20 beats per minute was seen in both the TRANSFORMS and FREEDOMS studies. This decrease was noted within 1–2 hours of the dose administration and persisted for approximately 6 hours. Only 0.8% in TRANSFORMS and 0.7% in FREEDOMS had symptomatic bradycardia. Two in the FREEDOMS group were treated for symptomatic bradycardia. First- or second-degree heart block was seen infrequently in both studies. No persistent or additional cardiovascular effects were seen with fingolimod with continued use. These first-dose cardiovascular effects have resulted in an FDA-mandated 6-hour observation period with the first dose of fingolimod.
Confirmed macular edema was diagnosed in 7 patients receiving fingolimod in TRANSFORMS and 6 in FREEDOMS.53,54 The majority of these cases occurred within 3 months of initiation of fingolimod and resolved with discontinuation of treatment with fingolimod. Visual acuity and retinal nerve fiber layer thickness in the fingolimod groups were similar to that seen in the IFN-β-1a and placebo groups in TRANSFORMS and FREEDOMS, respectively.
Skin cancers were diagnosed with greater frequency in fingolimod-treated groups than those treated with either interferon or placebo. Both in TRANSFORMS and FREEDOMS, all were local and were successfully treated with excision. Breast cancer was diagnosed more frequently in the fingolimod groups than in the interferon group in TRANSFORMS. 53 The incidence of breast cancer in FREEDOMS was higher in the placebo group than fingolimod groups. 54
Laboratory value abnormalities were seen with relative frequency in both phase 3 trials. Increased serum alanine aminotransferase (ALT) up to 3 times normal was the most frequently encountered laboratory abnormality. Lymphopenia relative to baseline measurements was seen in greater than 70% of those taking fingolimod in both TRANSFORMS and FREEDOMS as expected based upon fingolimod's proposed primary mechanism of action; true lymphopenia was rare. Lymphocyte counts returned to normal upon discontinuation of fingolimod. In TRANSFORMS, a 2%–3% decrease in mean forced expiratory volume in 1 second (FEV1) without alteration in lung volume or diffusion capacity. 53
There were 2 fatalities in the TRANSFORMS study: 1 from disseminated varicella zoster and 1 due to herpes simplex encephalitis. 53 There was 1 fatality in the FREEDOMS study due to suicide. 54 Similar numbers of herpes virus infections were seen among all 3 study groups. Infections more frequently identified in the fingolimod groups were urinary tract infection and lower respiratory tract infection.
In January 2012, the
Teriflunomide
Teriflunomide is the active metabolite of leflunomide, a drug FDA-approved for the treatment of rheumatoid arthritis. 57 Leflunomide is converted to teriflunomide by various cytochrome P450 isoenzymes. 58 Though leflunomide is an efficacious treatment for rheumatoid arthritis, it has potentially serious side effects such as interstitial lung disease and hepatotoxicity. Toxicity is thought to be related to ineffective enzymatic conversion of leflunomide to teriflunomide. Due to risks with leflunomide, the active metabolite, teriflunomide is being explored as a potential therapy for multiple sclerosis.
Teriflunomide's primary mechanism of action is inhibition of dihydro-orotate dehydrogenase (DHODH). 57 Dihydro-orotate dehydrogenase is necessary for the de novo synthesis of pyrimidines and, thus, DNA replication in rapidly proliferating cells such as lymphocytes. Because teriflunomide inhibits lymphocyte proliferation by interfering with DNA replication, its effect is cytostatic rather than cytotoxic. Other mechanisms of action for teriflunomide have been proposed based upon studies in the murine model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE). These other mechanisms include decreased production of interferon gamma, decreased T cell chemotaxis, and increased secretion of the anti-inflammatory cytokine, interleukin-10. 59
Clinical studies
O'Connor and colleagues recently published a positive 2-year, phase 3 study for teriflunomide, the teriflunomide MS oral (TEMSO) trial. 60 TEMSO included 1,088 patients with RRMS, SPMS, and progressive relapsing MS, who were blinded to treatment group and randomized 1:1:1 to either teriflunomide 7 mg, teriflunomide 14 mg, or placebo. The primary endpoint was ARR. Secondary endpoints were 3-month sustained progression of disability based upon a 1.0 point increase in EDSS score from baseline (for EDSS 5.5 and below) and the fatigue impact scale (FIS). MRI outcomes included total lesion volume, number of gadolinium-enhancing lesions, the volume of T1 hypointense lesions, number of unique active lesions (number of gadolinium-enhancing lesions or new or enlarged T2 lesions), and brain atrophy. In addition, cognitive function as measured by changes from baseline on the paced auditory serial addition test (PASAT-3) was a tertiary outcome measure in the extension study. 61
Both doses of teriflunomide had a significant positive impact on the primary endpoint of ARR, with a relative reduction in relapse rate compared to placebo of 31.2% and 31.5% for the 7 mg and 14 mg teriflunomide doses, respectively (
MRI outcomes in TEMSO.
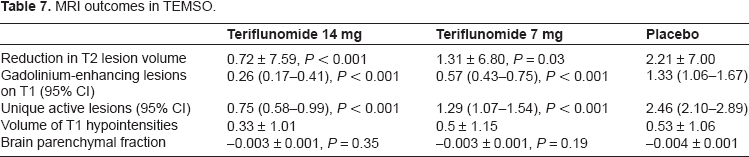
Preliminary results of a phase 3, non-inferiority trial comparing teriflunomide to Rebif® were released in December 2012 by Genzyme. 62 The TENERE trial was a 2-year, rater-blinded study comparing high dose (14 mg) and low dose (7 mg) teriflunomide to 3 times per week (tiw) intramuscular IFN-β-1a. In the trial, 324 participants were randomized 1:1:1 to 14 mg teriflunomide, 7 mg teriflunomide, or IFN-β-1a. The primary endpoint of the study, risk of treatment failure, was defined as confirmed MS relapse or discontinuation of study drug for any reason. There was no statistical difference among the 3 groups in risk of treatment failure. In addition, there was no statistically significant difference among high dose teriflunomide, low dose teriflunomide, and Rebif for the secondary endpoint of ARR (0.259, 0.410, 0.216, respectively). The complete results of the TENERE trial are expected to be published in late 2012.
Safety
Adverse events occurred with similar frequency in the teriflunomide groups and the placebo group in the TEMSO trial. 60 Common adverse effects in the TEMSO trial were diarrhea, nausea, hair thinning, and elevated alanine aminotransferase (ALT) levels. 60 Though a small number discontinued the study due to these symptoms, no participants discontinued as a result of persistent ALT elevation. Adverse events were similar in the TENERE trial, also including diarrhea and hair thinning as well as nasopharyngitis and back pain. 62 There was no significant difference in the frequency of adverse events among the teriflunomide and Rebif groups in the TENERE trial.
In the TEMSO trial, 1 malignant neoplasm was reported in the 14 mg teriflunomide group (cervical carcinoma) while 3 were seen in the placebo group in the initial trial. 60 In the extension arm, 2 malignancies occurred in high and low dose teriflunomide groups. 63 One basal cell carcinoma and 1 colon cancer were confirmed in the 7 mg group and 1 breast neoplasm and 1 renal cell cancer in the 14 mg group.
Adverse events occurring with higher frequency in the teriflunomide groups were increased blood pressure and skin problems. 60 Mean supine systolic and diastolic blood pressure elevation in the occurred in 5.4% of those in the low dose teriflunomide group, 5% in the high dose teriflunomide group, and 3.1% in the placebo group. Skin disorders including hypersensitivity without anaphylaxis occurred 10.3%, 11.2%, and 7.2% in the 7 mg, 14 mg, and placebo groups, respectively.
Pregnancy was reported as an adverse event in the TEMSO trial as teriflunomide's effect on pregnancy is unknown. 60 There were 11 pregnancies resulting in 4 spontaneous abortions, 6 induced abortions, and 1 healthy live birth. Though an increased rate of spontaneous abortion would be expected based upon teriflunomide's mechanism of action, the numbers from the TEMSO trial are small, preventing any conclusions with regard to its safety in pregnancy. The current recommendation is for an 18 to 24 month “washout” period or administration of 11 days of cholestyramine or activated charcoal prior to attempts at conception.
No serious opportunistic infections were seen in either teriflunomide group; however, there were 3 serious cases of pyelonephritis in the 14 mg teriflunomide group with 1 case prompting withdrawal from the study. Though no serious opportunistic infections were seen in the TEMSO trial, some concern exists for progressive multifocal leukoencephalopathy (PML) as 1 case has been reported in with leflunomide in a patient with systemic lupus erythematosus (SLE). 64 No cases of PML have been reported with use of teriflunomide.
BG-12
Fumaric acid esters have been used off-label for treatment of psoriasis in Europe for some time. A formulation of the FAEs dimethylfumarate and monoethylfumarate with the trade name, Fumaderm, was approved for treatment of psoriasis in 1994 in Germany. One of these FAEs, dimethylfumarate, is currently under investigation as a potential therapy for MS. Dimethylfumarate (DMF) is almost completely hydrolyzed in the small intestine to its active metabolite, monomethylfumarate (MMF). 65 Absorption of MMF is decreased by concurrent ingestion of food, though it remains highly bioavailable. 66 Its metabolism does not require the cytochrome p450 system, thus, few drug interactions would be expected. Monomethyl fumarate has a half-life of approximately 12 hours and the excretion of metabolites is primarily via respiration.
The mechanism of BG-12 in vivo is not fully understood. Its effects in vitro appear similar to those of other FAEs. Fumaric acid esters have been shown to shift the cellular cytokine profile from the pro-inflammatory Th1state to the less inflammatory Th2 state. 67 In humans and mice, this shift is in part due to stimulation of type 2 dendritic cell differentiation. Type 2 dendritic cells subsequently produce the anti-inflammatory cytokines, IL-10 and IL-4. 68 The shift from the Th1 to the Th2 state may also result in the apoptosis of activated T cells and decrease the expression of adhesion molecules, ICAM-1 and VCAM, protecting the CNS from influx of activated lymphocytes. It has also been shown to decrease the antigen-presenting ability of monocytes and macrophages. 69
Clinical studies
Prior to studies with BG-12, the FAE formulation, Fumaderm®, was studied in MS. Fumaderm's effect on MS was initially explored in a 4-phase, open-label trial in 10 patient with RRMS.
67
The 4 phases were a 6-week baseline, an 18-week treatment arm with 720 mg of Fumaderm daily, a 4-week washout, and a 48-week treatment arm with 360 mg of Fumaderm daily. The primary outcome measures were the number and volume of gadolinium-enhancing lesions. Exploratory outcomes included change from baseline in EDSS score, in ambulation index (AI), and in nine-hole peg test (9-HPT). At conclusion of the 70-week trial, the mean number of gadolinium-enhancing lesions had decreased from 11.28 to 0.28 (
Exploratory outcomes in Fumaderm trial.

The phase 2 trial examined the effect of 2 different doses and dosing regimens of dimethylfumarate (BG-12), 120 mg once daily, 120 mg three times daily (tid), or 240 mg tid, versus placebo in 257 patients with RRMS in a 1:1:1:1 randomized, double-blind design. 70 The primary outcome variable was total number of new gadolinium-enhancing lesions over 4 MRI scans at specified time points. Secondary MRI endpoints were cumulative number of new gadolinium-enhancing lesions, number of new or enlarging T2-hyperintense lesions, and new T1-hypointense lesions. Other endpoints assessed were number of relapses, disability progression, and the safety of BG-12.
The high dose BG-12 group showed a significant decrease in mean number of new gadolinium-enhancing lesions (
The first completed phase 3 study (DEFINE) of BG-12 compared 240 mg of BG-12 twice daily (bid) and 240 mg tid to placebo in a randomized, double-blind trial.
71
One thousand thirty-four patients with RRMS were randomized 1:1:1 to bid BG-12, tid BG-12, and placebo. The primary endpoint was the proportion of patients relapsing at 2 years. Secondary endpoints were ARR and sustained disability progression as measured by EDSS. BG-12 met all primary and secondary endpoints specified in the study. Both groups of BG-12 showed a reduction in the risk of relapse. The risk of relapse was decreased by 49% in the bid dosing group and by 50% in the tid dosing group (
The CONFIRM trial was the second positive phase 3 trial for BG-12. 73 It was a double-blind, placebo-controlled trial with a reference comparator arm. CONFIRM examined 1,430 patients with RRMS randomized 1:1:1:1 to BG-12 240 mg bid, 240 mg tid, GA, or placebo. The primary endpoint was annualized relapse rate at 2 years. Secondary endpoints were number of new orenlarging T2-hyperintense lesions, new T1-hypointense lesions, proportion of patients who relapsed, and 3-month sustained disability progression as assessed by the EDSS. BG-12 and GA were not directly compared in the CONFIRM trial, as the study was not powered to make such a comparison.
BG-12 met both primary and secondary endpoints in the CONFIRM trial.
73
Annualized relapse rate was reduced by 44% for bid and 51% for tid dosing of BG-12 (
Safety
As Fumaderm® has been approved in Germany for treatment of psoriasis since 1994, the adverse effects of Fumaderm® are fairly well known. The most common effect of BG-12 is episodic flushing, which decreases in frequency with continued administration of BG-12. 70 These episodes usually begin within 30 minutes of taking the medication and resolve in about 90 minutes. Flushing occurred in about 1/3 of patients in the phase II trials and in the DEFINE phase III trial of BG-12. 70 Gastrointestinal (GI) upset including nausea, diarrhea, and upper abdominal pain was seen in up to 20% of patients in the phase II trial. 70 Rarely, BG-12 caused elevation of transaminases up to 3 times normal; however, this resolved with discontinuation of the medication. Adverse effects of BG-12 appear to be dose-related.
Serious adverse effects of BG-12 were infrequent in the phase II study and the most frequent serious adverse event reported was multiple sclerosis relapse. 70 With BG-12 treatment of psoriasis, cases of acute renal failure have been reported, though other literature seems to refute this risk. 65 No serious infections or neoplasia have occurred with any frequency with FAE treatment.
Laquinimod
Laquinimod is a structural cousin of linomide, which has been tested for efficacy in MS. 74 Phase 3 trials of linomide were prematurely discontinued due to an unacceptably high rate of cardiac and other serious systemic adverse effects. An extensive investigation on the structure-activity relationships of linomide that may be responsible for its adverse effects was undertaken. This resulted in 60 compounds related to linomide with potential for use in autoimmune disease. From these 60 compounds, laquinimod was selected based upon its absence of pro-inflammatory effects in animal models. Laquinimod is chemically and pharmacologically distinct from linomide, thus, improving its safety profile versus linomide. 75
Several potential mechanisms of action have been proposed for laquinimod. In EAE, laquinimod has been shown to decrease the influx of lymphocytes into the CNS and decrease the concentration of macrophages and T cells in the spinal cord. 76 Also in the EAE model, laquinimod protects axonal loss by an unclear mechanism and causes a shift from the pro-inflammatory Th1state to the anti-inflammatory Th2 state. Lastly, in the EAE murine model, laquinimod may increase CNS and serum levels of brain neurotrophic growth factor. Laquinimod is well-absorbed following oral administration and is highly bioavailable (up to 95%). 77 It has an extremely long half-life of 80 hours. It does not readily cross the blood-brain barrier. Laquinimod is extensively metabolized in the liver to several intermediary metabolites by the cytochrome P450 system, specifically the CYP3A4 enzyme. 78 Drug-drug interactions may be seen as CYP3A4 is the enzyme primarily responsible for the metabolism of common medications such as anti-depressants and antibiotics.
Clinical studies
Two phase 3 clinical trials of laquinimod in RRMS, the ALLEGRO and the BRAVO trials, have been completed. The ALLEGRO trial was a 2-year, randomized, double-blind, placebo-controlled trial with 1,106 RRMS patients. 79 Patients were randomized to 1:1 laquinimod 0.6 mg or placebo. The primary endpoint of the study was ARR. Secondary MRI endpoints measures were total number of gadolinium-enhancing lesions and new or enlarging T2 lesions. The secondary clinical endpoint was 3-month confirmed disability progression based on EDSS and MSFC scores. Other exploratory endpoints were new T1 hypointense lesions, brain atrophy, and the number of relapses requiring hospitalization or IV steroids.79,80
The primary endpoint of the study, ARR, showed positive results for laquinimod. There was a 23% lower ARR than the placebo group (
BRAVO was randomized and double-blinded but compared laquinimod to both placebo and a reference arm of weekly intramuscular interferon β-1a.
82
The primary endpoint of reduction of ARR did not differ significantly for laquinimod. After an adjustment pre-specified in the study protocol was performed for factors suggesting less aggressive disease at baseline in the placebo group, the adjusted ARR was reduced in the laquinimod group to 0.29 versus 0.37 in the placebo group (
Teva Pharmaceuticals, the developers of laquinimod, chose not to file for FDA approval following the results of the BRAVO trial. Though laquinimod appears to have similar efficacy in reducing ARR as currently available first-line therapies, it reduced disability progression and brain atrophy in both the ALLEGRO and BRAVO trials. This data suggests that laquinimod may be neuro-protective, thus, making it an attractive candidate for future study in progressive forms of MS.
Safety
As compared to its predecessor, linomide, laquinimod has far superior safety and tolerability. In the ALLEGRO trial, there was no significant difference in incidence of adverse effects. 79 This was consistent with the results of the phase 2 clinical trials. There were no consistently seen serious adverse events with laquinimod in either phase 2 trial.74,83 Common adverse effects included back pain and arthralgias (about 5%). In the extension of the phase 2 trial by Comi et al, elevation of serum ALT resulted in early termination of the study by 3 participants. 83 There were no other associated signs or symptoms of hepatic dysfunction. Changes in other laboratory values were clinically insignificant. Viral infections, specifically, herpes simplex and herpes zoster were seen more frequently in the Comi et al phase 2 study; however, all infections were cutaneous and self-limited. There were no adverse effects reminiscent of linomide in any of the trials of laquinimod.74,83,84
Conclusion
The therapeutic pipeline for MS is enriched with novel agents that have the potential to provide improved efficacy and ease of administration for patients. Selection of an appropriate disease modifying therapy, however, will become increasingly more complex as more therapies become available. The risk to benefit ratio of a given therapy must be carefully considered in each individual patient, in addition to the order in which potential future therapies are offered.
An escalation model beginning with so-called “first line” disease modifying therapies would be utilized first. These have been proven efficacious and have highly favorable side effect profiles. The current injectable therapies (IFN-β-1a, IFN-β-1b, and GA) would likely remain the preferred agents, given the almost two decades of global experience with these medications. One may consider adding BG-12 to this list of first line agents. Patients who experience unacceptable breakthrough disease or intolerable adverse effects on a given first line therapy would be transitioned to another first line therapy with a different mechanism of action (eg, GA to IFN-β or vice versa) or they would escalate to so-called “second line” treatments that may have improved efficacy, but less attractive safety profiles. These second line therapies currently include natalizumab and fingolimod, and may soon include alemtuzumab, ocrelizumab and ofatumumab. In patients with more aggressive disease, an alternative approach would be to consider an induction model, where a therapy with strong efficacy but strong safety concerns would be given first, followed by a “maintenance” therapy. An example of such a model might include initial treatment with alemtuzumab, followed by IFN-β, GA, or BG-12.
Clearly, neurologists will need to carefully match each patient's disease profile with an appropriate therapy while carefully weighing the risks and the benefits of these newer agents. The development of biomarkers to help prognosticate a given patient's likely disease severity and to help predict treatment response
Author Contributions
Wrote the first-draft of the manuscipt: JN, BMF. Contributed to the writing of the manuscipt: JN, BMF, AB, MKR. Agree with manuscript results and conclusions: JN, BMF, AB, MKR, DP. Jointly developed the structure and arguments for the paper: JN, BMF, AB, MKR, DP. Made critical revisions and approved the final version: JN, BMF, AB, MKR, DP. All authors reviewed and approved the final manuscript.
Funding
Author should state fund sources here. If none exist this section may be deleted. This work was supported by a Sylvia Lawry Physician Fellowship Award (J. Nicholas) and a Multiple Sclerosis Clinical Care Physician Fellowship Award (B. Morgan-Followell) from the National Multiple Sclerosis Society.
Competing Interests
JN and BNF received grant support from the National Multiple Sclerosis Society. MKR has acted as a consultant to Biogen-Idec, Diogenix, EMD Serono, Elan and Novartis. Authors have previously received NIH grants.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest. Provenance: the authors were invited to submit this paper.
Footnotes
Acknowledgement
This work was supported by a Sylvia Lawry Physician Fellowship Award (J. Nicholas) and a Multiple Sclerosis Clinical Care Physician Fellowship Award (B. Morgan-Followell) from the National Multiple Sclerosis Society.