
Abstract
Using a philosophical approach or deductive reasoning, we challenge the dominant clinico-radiological worldview that defines multiple sclerosis (MS) as a focal inflammatory disease of the central nervous system (CNS). We provide a range of evidence to argue that the ‘real MS’ is in fact driven primarily by a smouldering pathological disease process. In natural history studies and clinical trials, relapses and focal activity revealed by magnetic resonance imaging (MRI) in MS patients on placebo or on disease-modifying therapies (DMTs) were found to be poor predictors of long-term disease evolution and were dissociated from disability outcomes. In addition, the progressive accumulation of disability in MS can occur independently of relapse activity from early in the disease course. This scenario is underpinned by a more diffuse smouldering pathological process that may affect the entire CNS. Many putative pathological drivers of smouldering MS can be potentially modified by specific therapeutic strategies, an approach that may have major implications for the management of MS patients. We hypothesise that therapeutically targeting a state of ‘no evident inflammatory disease activity’ (NEIDA) cannot sufficiently prevent disability accumulation in MS, meaning that treatment should also focus on other brain and spinal cord pathological processes contributing to the slow loss of neurological function. This should also be complemented with a holistic approach to the management of other systemic disease processes that have been shown to worsen MS outcomes.
Keywords
Introduction
A large proportion of people with multiple sclerosis (MS) continue to experience clinical deterioration despite a lack of overt ongoing inflammatory disease activity. To this end, such patients exhibit disability progression despite being relapse-free and exhibiting neither contrast-enhancing T1-weighted (T1w) lesions nor new or enlarging T2-weighted (T2w) lesions on magnetic resonance imaging (MRI). This is often referred to as progression independent of relapse activity (PIRA) or smouldering MS, 1 which is distinct from relapse-associated worsening (RAW; see Figure 1 for definitions). Based on a synthesis of pathology, neuroimaging, and clinical data, we propose that the ‘real MS’ is likely to be driven by a primary smouldering process accompanied by a superimposed inflammatory activity that potentially represents the host immune response to underlying causes of the disease. 2 Overwhelming evidence from MRI and pathological studies indicate that the progressive neuroaxonal loss that underpins the accumulation of unremitting disability is present from the very early stages of the disease.3–6 From a biological perspective, this would imply a continuum between the relapsing and progressive stages of MS, which are distinguished only by quantitative rather than qualitative pathological differences. 7 What we see clinically is an interplay between the effects of focal inflammatory events superimposed on the nervous system, which can be functionally compromised depending on the extent of previous pathological insults, the brain’s reserve capacity, and its ability to recover function or to compensate for damage incurred. 8

Relapse-Associated Worsening (RAW) and Composite Progression Independent of Relapse Activity (PIRA) Definitions. This figure is based on Kappos et al. 1 and is a schematic representation of RAW and PIRA, which are non-mutually exclusive drivers of confirmed disability accumulation (CDA) in both relapsing and progressive forms of MS. The baseline is the reference point for disability changes measured over time; in the context of clinical trials, this is the time of randomisation to study treatment, but in the context of the clinic, this would be the reference disability assessment visit from which subsequent changes are measured over time. The shaded areas represent the intervals around the neurological assessments that had to remain free of relapses to fulfil the criterion of independence from relapses (at initial event and confirmation points). Neurological assessments were scheduled to occur every 12 weeks, according to the protocol of the study; if a relapse occurred, there was one neurological assessment outside of the schedule, at a point corresponding to the leftmost point on the relapse triangle. EDSS, Expanded Disability Status Scale; IID, initially assessed increase of disability; MS, multiple sclerosis.
This review will provide a biological perspective of the pathological drivers responsible for smouldering MS. We will discuss how the smouldering process can be assessed in routine clinical practice, and how, as a result of these insights, unmet therapeutic needs in MS can be addressed. A shift away from simply targeting relapses and focal MRI activity will be proposed, with the focus of attention redirected to halting the putative processes responsible for smouldering MS.1,8 Therapeutically targeting these processes will have major implications for future clinical trial design. In addition, we will almost certainly require either dual-action or combination therapies to address more broadly the different pathological mechanisms driving smouldering MS.
Case study
A 39-year-old male with established secondary progressive MS (SPMS) and an Expanded Disability Status Scale (EDSS) score of 6.0 asks if he should switch his treatment from fingolimod to other disease-modifying therapies (DMT) in order to halt his disease progression. Over the past 3 years, his EDSS score has increased from 4.0 to 6.0, but he has had no superimposed relapses or new MRI lesions. Based on the Lublin classification 9 (Table 1 and Figure 2), this patient has inactive SPMS and, therefore, would not be eligible for switching to another DMT. The patient asks, ‘How can I have “inactive disease” when my legs are getting weaker?’; he now needs a walking stick, which he did not require 3 years ago.
This scenario is seen frequently by neurologists and makes up a significant proportion of patients with advanced MS. Despite the very effective suppression of focal inflammatory disease activity, patients often continue to experience a progressive accumulation of disability. Arguably, this suggests that anti-inflammatory DMTs are simply converting patients with relapsing forms of MS into a phenotype that is very reminiscent of what is seen in primary progressive MS (PPMS).

MS, multiple sclerosis; PPMS, primary progressive multiple sclerosis; SPMS, secondary progressive multiple sclerosis.
Can be another time frame, as long as this is specified.
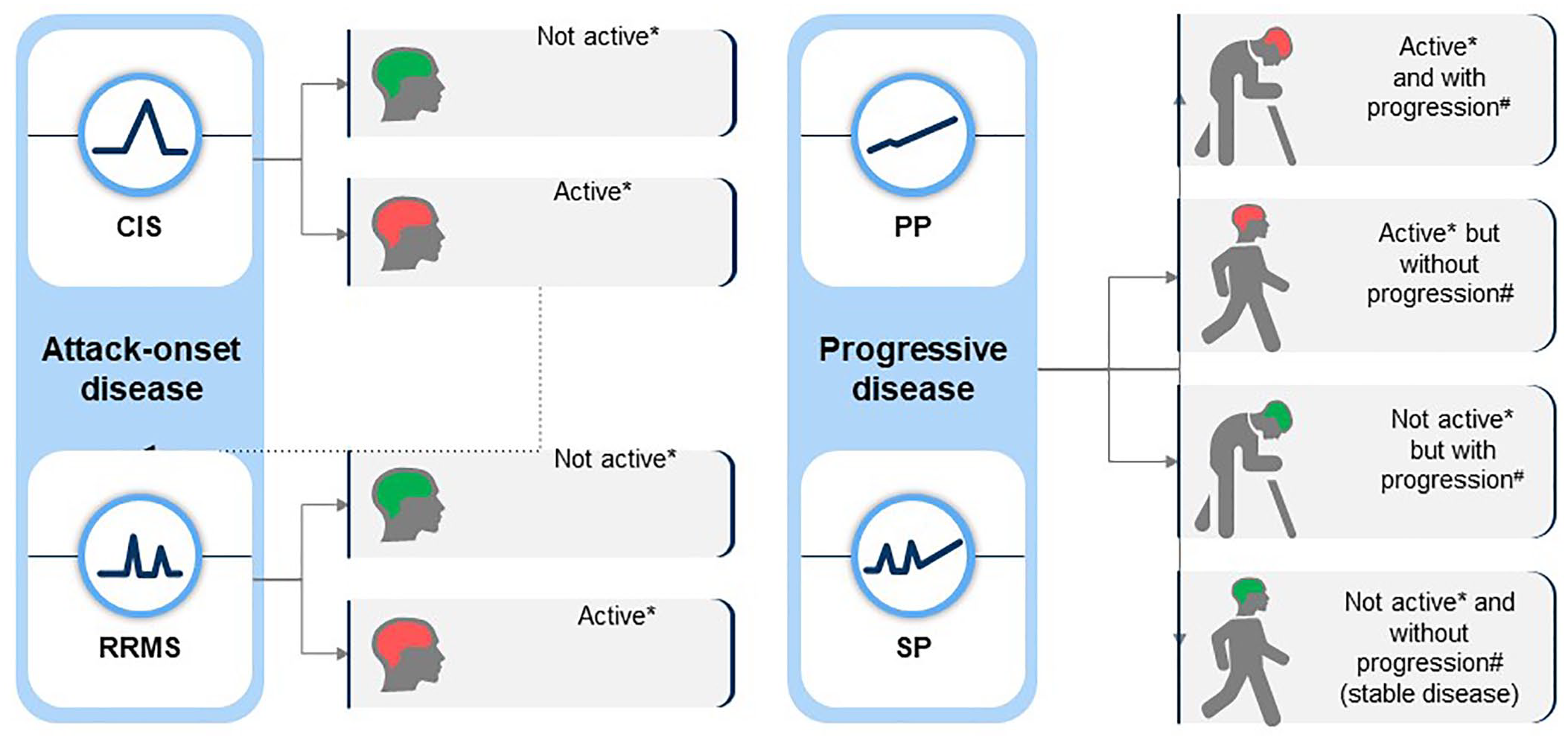
Lublin 2013 multiple sclerosis phenotype descriptions for relapsing and progressive disease (based on Lublin et al. 9 ).
Contemporary MS pathogenesis, natural history, and classification system
Autoimmune (outside-in) versus neurodegenerative (inside-out) hypothesis
Based on the current dogma, MS is considered primarily an outside-in disease triggered by T cell-mediated autoimmune peripheral events. Therapeutically targeting peripheral immunological events by using either immunodepleting agents, for example, alemtuzumab or haematopoietic stem cell transplantation, or lymphocyte anti-trafficking agents such as natalizumab, proves this hypothesis. However, while both strategies are effective in shutting down most of the focal inflammatory events, they do not necessarily stop disease worsening.1,12 Indeed, in relapsing-remitting MS (RRMS), the effective therapeutic suppression of relapses does not always correlate with the prevention of long-term disability accumulation, thus highlighting a disconnect between mechanisms underlying inflammatory attacks and those responsible for disease progression.1,12 The biology of smouldering MS, which will be discussed later in this paper, argues in favour of the hypothesis that MS is primarily an inside-out disease that starts in the central nervous system (CNS), and that focal inflammatory events are an epiphenomenon to the primary neuroaxonal loss. This scenario potentially promotes the release of antigenic myelin fragments, thereby triggering host innate and adaptive immune responses.2,13
Pathology
The concept that MS is a two-staged disease, initially dominated by an inflammatory relapsing phase that transitions into a non-inflammatory neurodegenerative phase, has been largely based on clinical and MRI observations.12,14 However, in contrast to this view, pathological studies show that active ongoing inflammation and demyelination in the CNS can be observed even in the terminal or end stage of MS. 15 In a large study that analysed 7562 brain lesions from 182 MS post-mortem cases with a mean disease duration of 29 years, 57% of the detected MS lesions were classified as chronic active lesions. 15 Importantly, active or chronic active lesions were found in 78% of the cases, with similar levels of lesion activity seen in SPMS and PPMS subgroups. 15
Indeed, relapses overlapping the primary and secondary progressive course can be observed in up to 40% of patients16,17 and were reported during the PPMS ocrelizumab trial (ORATORIO study) in 11% and 5% of the placebo and treatment groups, respectively. 18 Notably, 27% of that trial’s subjects had inflammatory activity on their baseline MRI. 18 Furthermore, in more advanced PPMS, relapses and focal MRI activity were observed after the discontinuation of DMTs.19,20
Overall, these observations concur with pathology studies showing, both in SPMS and PPMS cases, similar degrees of inflammatory infiltrates, axonal loss, and cortical demyelination, thus supporting the notion of MS being a single-stage disorder.21–24 This is in line with epidemiological observations of a unified disease model, where PPMS and SPMS patients manifest clinical progression at similar mean ages and experience a similar rate of disability accumulation.17,25 The clinical features of MS appear to be mostly age-dependent. Irrespective of the disease duration, the MS clinical phenotype becomes less inflammatory with age, with decreasing numbers of relapses and new MRI lesions, while the risk of developing progressive MS increases proportionally. 26 However, it has previously been shown that the risk of SPMS reaches a maximum and then decreases in older ages,27,28 indicating a role for ongoing inflammation in smouldering MS. The reason why relapses and focal MRI activity become less frequent with age and/or disease duration may have something to do with as yet undetermined qualitative immunological changes. This would imply that the immune response to the primary insult within the CNS, and its clinical correlates, may differ with age. These observations also challenge the Lublin classification, which implies by the use of distinct categories and one-way arrows, that once the MS phenotype becomes progressive and non-relapsing, it cannot revert to a relapsing phenotype 9 (see Figure 2). 17
End-organ damage
People with MS exhibit an accelerated brain volume loss that correlates with cognitive impairment and long-term disability.29,30 Demyelination, neuroaxonal and synaptic loss are the pathological substrates underpinning brain and spinal cord volume loss.31,32 However, whole-brain volume measurements are confounded by physiological factors that include circadian fluid shift, inflammatory oedema, and gliosis, as well as superimposed age-related and non-related changes. Therefore, the use of brain atrophy as a biomarker at an individual patient level can be challenging, whereas at a group level, whole-brain volume changes are considered an integrator of end-organ damage and are predictive of the clinical outcome.33,34 With the emerging use of new regional brain volume measurement techniques and advanced neuroimaging techniques, 35 we believe the measurement of volume changes will be gradually incorporated into clinical practice to assist with therapeutic decision-making and in the management of individual patients, thus contributing to individualised patient care (see below). 34
Pathological drivers of smouldering or ‘real’ MS
Numerous mechanisms have been proposed to drive smouldering MS (see Figure 3), several of which are arguably downstream of focal inflammatory lesions, while others may be independent of focal inflammatory lesions and be the cause of MS.

The pathological drivers of smouldering MS. Apart from acute focal damage characterised by axonal transection and conduction block that occurs over days to weeks and causes relapse-associated worsening (RAW), there are delayed time-dependent processes that are responsible for smouldering MS. Demyelination and energy deficits are responsible for delayed worsening, which occurs over weeks to months. Whether this is dependent or independent of ongoing focal inflammation is a moot point. This is then followed by post-inflammatory neurodegenerative processes, which run their course over many years and include microglial and innate immune mediators, ongoing energy deficits, antibody-mediated damage, and possible viral infection. Finally, age-related neurodegenerative processes that are premature and accelerated are responsible for late-onset disability progression, which plays out over decades.
Acute axonal and synaptic loss
It has been shown that the acute clinical deficit associated with new focal inflammatory MS lesions is correlated with axonal transection, subsequent Wallerian degeneration, and a downstream loss of synapses. 4 ,36–38 Proximal dying back of axons and associated neuronal loss have been well described in optic neuritis 39 and are likely to occur in other central pathways. It remains to be determined whether the axonal regrowth to restore lost function occurs in MS lesions 38 similarly to that observed in animals and in the human peripheral nervous system; for example, in poliomyelitis, the sprouting of surviving axons contributes to repair following a pathological insult. 40 Therefore, it is plausible to hypothesise that axonal sprouting primes neurons to die off prematurely as a result of an excessive metabolic burden placed on the surviving axons. 40
Demyelination
Demyelination and acute conduction block are other factors contributing to focal deficits within inflammatory lesions. 41 Axonal plasticity or the synthesis and insertion of sodium channels along demyelinated axonal segments restores nerve conduction, albeit at a much lower velocity and with a higher energy requirement. 42 These demyelinated axonal segments have a reduced safety factor of conduction, making them more susceptible to increased temperature or fatigue, and thus accounting for intermittent but reversible symptoms such as Uhthoff’s phenomenon. 42 In addition, demyelinated axons are less resilient and more likely to die prematurely due to an excessive metabolic burden or from axonal excitotoxicity stemming from the proinflammatory microenvironment in MS lesions. Although remyelination is well documented in MS, it is incomplete and fails with age. 43 As demyelinated axons are more susceptible to degeneration, the failure of remyelination in MS is likely to be one of the contributing factors to smouldering MS. 44
Macrophage/microglial activation
Activation of microglia and recruited macrophages are found in acute MS lesions and persists in chronic active and inactive MS lesions. Activated microglia produce proinflammatory cytokines and inflammatory mediators, which are hypothesised to drive both acute and chronic axonal loss. Focal smouldering inflammatory activity has been reported in several MS lesion types, encompassing chronic active/smouldering lesions and subpial cortical lesions. At autopsy, 20–40% of white matter lesions are categorised as slowly evolving lesions (SELs). 24 These are characterised by a low degree of inflammation and with T and B cells at the lesion core, a dense network of activated iron-laden microglia/macrophages expressing the pro-inflammatory markers CD68 and p22phox in a glial wall, and by proliferating oligodendrocytes at the lesion edge. 15 ,45–48 Microglial activation may also contribute to the failure of oligodendrocytes to remyelinate neurons, an effect that may be dependent on the stage of the lesions. 49
In contrast to other MS lesions, which have a tendency to shrink in gliotic stages, SELs with paramagnetic rims of activated microglia at their edges contribute to the failure of remyelination, resulting in further destruction of the surrounding parenchyma. 50 The presence of SELs has been correlated with a more severe clinical outcome. 50 In addition, patients with heightened microglial activation, as determined by the expression of 18 kDa translocator protein (TSPO) binding to the radioligand PK11195 on positron emission tomography (PET) imaging, are more likely to experience PIRA. 51 This observation suggests that TSPO brain PET ligands are good markers of smouldering MS. A longitudinal PET study demonstrated a decrease, but not suppression, in microglial activation in natalizumab-treated SPMS patients compared with untreated patients. 52 Importantly, this decrease was associated with slower disability progression during 4 years of follow-up. 52 This implies that trafficking of peripheral immune cells into the CNS contributes to, but does not drive, smouldering MS.
It remains to be established if pharmacologically reducing microglial activation will translate into improved long-term outcomes with established DMTs. CNS-penetrant Bruton’s Tyrosine Kinase inhibitors (BTKi), which are known to inhibit microglial activation through the Fc-gamma receptor, hold promise as a means to prevent disease progression unrelated to relapses.53,54 This, however, must be balanced by the potential role that microglia play in clearing up debris in the CNS and in promoting remyelination. 55 Interestingly, BTKi was recently shown to promote repair. 54 Therefore, the qualitative shift in microglia from a proinflammatory to a pro-remyelinating phenotype may prove to play a key role in controlling smouldering MS.
Chronic oxidative injury
Oxidative stress and damage were shown to be more severe in the brains of patients with progressive, compared with relapsing MS. 56 Nitric oxide and its metabolites, superoxide dismutase, catalase, glutathione reductase, inducible nitric oxide synthase, protein carbonyl, 3-nitrotyrosine, isoprostanes, malondialdehyde and products of DNA oxidation have been identified in MS lesions and are potential therapeutic targets for addressing mechanisms underlying smouldering MS. 57 After a positive clinical trial in progressive MS, 58 alpha-lipoic acid and other antioxidants are considered to be some of the most promising add-on therapies to prevent the accumulation of progressive disability in MS. 59 Lipoate and its reduced form dihydrolipoate react with reactive oxygen species and protect membranes by interacting with vitamin C and glutathione. 60 Lipoate also functions as a redox regulator of several proteins, including the proinflammatory transcription factor NF-kappa B and is therefore thought to also have anti-inflammatory effects. 60
Age-related iron accumulation
Age-related iron accumulation occurs physiologically in the human brain, reaching a plateau between 40 and 50 years of age. 61 Accumulating iron, which is known to be increased in MS brains, is released from damaged oligodendrocytes and myelin during active demyelination. 62 The degree of neuroaxonal loss is more extensive in brain areas with the highest iron content, in particular the deep grey matter nuclei. 63 Iron can promote the production of reactive oxygen species and enhance the production of proinflammatory cytokines. Therefore, iron chelation therapy and the stabilisation of hypoxia-inducible transcription factor 1α, which is normally upregulated under hypoxic conditions, have been proposed as potential neuroprotection strategies in MS. Pilot studies of the iron chelator deferoxamine have been undertaken in MS,64,65 but no follow-on efficacy trials have been attempted.
Mitochondrial damage and energy deficits
The accumulation of mitochondrial damage in MS lesions was shown to contribute to axonal loss by inducing ‘virtual hypoxia’. 66 The mitochondrial injury occurs as a consequence of oxidative stress and in some MS lesions defects in mitochondrial respiratory chain complex IV have been described, thus explaining the hypoxic tissue injury secondary to energy deficiency. 67 Altered mitochondrial function in axons might be of particular importance, as this may lead to chronic axonal stress and an imbalance in ion homoeostasis, which can result in neuronal death. 68
It has been hypothesised that a deficiency of biotin in MS might result in energy deficits, 69 where a reduced flux of biotin through the tricarboxylic acid cycle results in lower ATP production. This occurs as a consequence of lower biotin-dependent pyruvate carboxylase activity as well as diminished flow through the mitochondrial electron transport chain due to deficient cytochrome c oxidase or complex IV activity. 70 Early phase 2 trials suggested that high-dose biotin improved disability in a small proportion of subjects with progressive MS, 69 but this was not replicated in a large multicentre phase 3 study. 71 Animal study results 72 suggested that oxygen could serve as a possible therapeutic option for addressing mitochondrial damage in MS. 73 However, hyperbaric oxygen provided no benefit to progressive MS patients. 74 A caveat to these observations is that the studies were carried out when progressive MS trial designs were still being developed and studies were clearly underpowered. Therefore, proponents of the MS energy deficit hypothesis want to revisit the use of oxygen as a treatment for MS, particularly as a neuroprotective strategy. 72
Infections
Infections alter the natural history of MS as the probability of relapses increases in the 5- to 6-week ‘at-risk’ period after infection.75–78 These observations were largely described in the pre-DMT era and do not apply to patients on treatments. In more advanced MS, the occurrence of infections may transiently worsen pre-existing symptoms because of concomitant fever, which causes reversible conduction block or Uhthoff’s phenomenon. 79 Interestingly, a systemic infection may upregulate innate immune responses in the CNS due to the endocrine effect of cytokines. This has been described to accelerate neuroaxonal loss in animal models of MS 80 and may also occur in MS patients. 76
Although many people believe that Epstein–Barr Virus (EBV) is the cause of MS, experimental proof is lacking. It has been suggested that persistent EBV infection of the CNS drives ongoing MS disease activity.81,82 EBV and other herpes viruses transactivate human endogenous retroviruses (HERVs), which are upregulated in the brains of patients with MS. 83 The production of envelope (Env) proteins from HERV-W and HERV-K induce proinflammatory and superantigen (SAg)-like effects, 84 which may be relevant to the pathological processes that drive smouldering MS lesions. These and other observations support the rationale for exploring the potential therapeutic effects of antiviral agents in MS.
Interestingly, all licensed MS DMTs target and/or reduce absolute numbers of memory B cells, 85 with the exception of natalizumab that increases peripheral counts but blocks their trafficking into the CNS.86,87 The memory B cells contain a subset of cells that are latently infected with EBV. 85 In addition, beta-interferon and teriflunomide 86 have direct, broad antiviral effects that may be relevant to their therapeutic mode of action in MS.
The paradox between focal inflammatory activity (relapses / MRI) and disability progression
One of the MRI-clinical paradoxes that we wish to consider refers to the observation that focal MRI activity, in the form of contrast-enhancing T1w and/or new/enlarging T2w lesions, and clinical relapses do not predict long-term disability outcomes in natural history studies of MS, nor in patients on placebo in clinical trials. Epidemiological observations indicate that a high frequency of early relapses is associated with faster disease evolution.88–91 However, late relapses 88 and inflammatory attacks overlapping the progressive course16,92 do not influence disability accumulation. Even among patients with a high number of early relapses (⩾3) within the first 2 years of the disease, a proportion can experience a mild disease course in the long term. 93 Further evidence of a disconnect between the occurrence of relapses and late disability is provided by long-term follow-up studies of patient cohorts in pivotal DMT clinical trials. Despite treatment of patients with interferon-beta (evident-disease activity), a larger number of early relapses was associated with poor long-term outcomes; in contrast, in subjects initially randomised to placebo, the frequency of early attacks had no predictive value. 94 Similarly, in a large real-world data set of 2,466 patients followed for at least 10 years, on-therapy relapses carried greater weight than off-therapy relapses in predicting disability progression. 95 If focal inflammation detected clinically as relapses or radiologically as new lesions were the primary driver of the MS progression, then inflammatory attacks and MRI activity would be expected to predict long-term outcomes both in untreated patients and in those on a DMT.
By applying Prentice Criteria for a surrogate endpoint 96 (see Table 2) and deductive reasoning, relapses and/or focal MRI activity cannot be validated as surrogate endpoints for MS progression and are therefore likely to be an epiphenomenon to the pathological processes driving the disease. We propose that focal inflammation occurs in response to the cause of MS; that is, the real disease. The focal inflammatory activity (relapses and/or MRI activity) does not predict disability outcomes in untreated people and is frequently referred to as the clinico-radiological paradox, 97 while in patients on a DMT, it represents a marker that the therapy is not affecting the underlying primary driver of the MS disease process.
Prentice Criteria for a surrogate endpoint. 96

Another example of this paradox concerns the recent observation that patients with increasing ocrelizumab exposure have greater levels of B-cell depletion in their peripheral blood. 98 It was shown that following the administration of ocrelizumab 600 mg every 6 months, serum concentrations of the drug were higher among subjects weighing < 60 kg and lower among subjects weighing > 90 kg, compared with subjects weighing 60–90 kg. 98 This dose effect on B-cell depletion was not reflected in the reduction of relapses or of focal lesions on MRI, as all levels of ocrelizumab exposures were associated with almost complete suppression of focal inflammatory activity. However, a clear dose effect was observed, with more significant prevention of disability progression in both relapsing and PPMS patients having higher levels of peripheral B-cell depletion. 99 This further supports a disconnect between the pathological processes driving focal inflammation and those responsible for non-relapsing disability progression. 99 As a result of these observations, ocrelizumab doses of 1200 mg or 1800 mg every 6 months are being compared with the licensed dose of 600 mg every 6 months in two clinical trials (ClinicalTrials.gov Identifiers: NCT04117529 and NCT04548999). Whether the higher ocrelizumab exposure is targeting peripheral deep tissue B cells or intrathecal B cells is a moot point, but these observations highlight the need for targeting mechanisms driving smouldering MS beyond focal inflammatory events.
Interestingly, ofatumumab, a fully humanised monoclonal anti-CD20 therapy, was shown to be superior to teriflunomide in suppressing relapses and focal inflammatory MRI activity, but lacking a greater impact on slowing the rate of brain atrophy over 2 years. 100 Therefore, despite its relatively modest impact on focal inflammation, teriflunomide successfully impacts the primary MS pathology by reducing brain volume loss.101,102 These observations further support a disconnection between the systemically driven inflammation and the smouldering pathology occurring in the end-organ or target organ. In addition, teriflunomide is more effective when used as a second or third-line agent, 103 raising the question of whether its undetermined mode of action targets mechanisms that are downstream of inflammation or the processes driving smouldering MS, for example, by inhibiting microglial responses. 104
Advanced imaging
Advanced imaging techniques, which can be defined as imaging modalities that are not yet available in routine clinical practice, provide an opportunity to assess the microscopic features of MS. For example, pathology studies have shown that low-level chronic inflammatory activity in microglial cells is also present in normal-appearing white and grey matter. 105 The appearance of clusters of microglia precedes in some cases the formation of microglial lesions106–109 (see Figure 4). Advanced imaging can be used to investigate such changes in the normal-appearing white matter (NAWM).
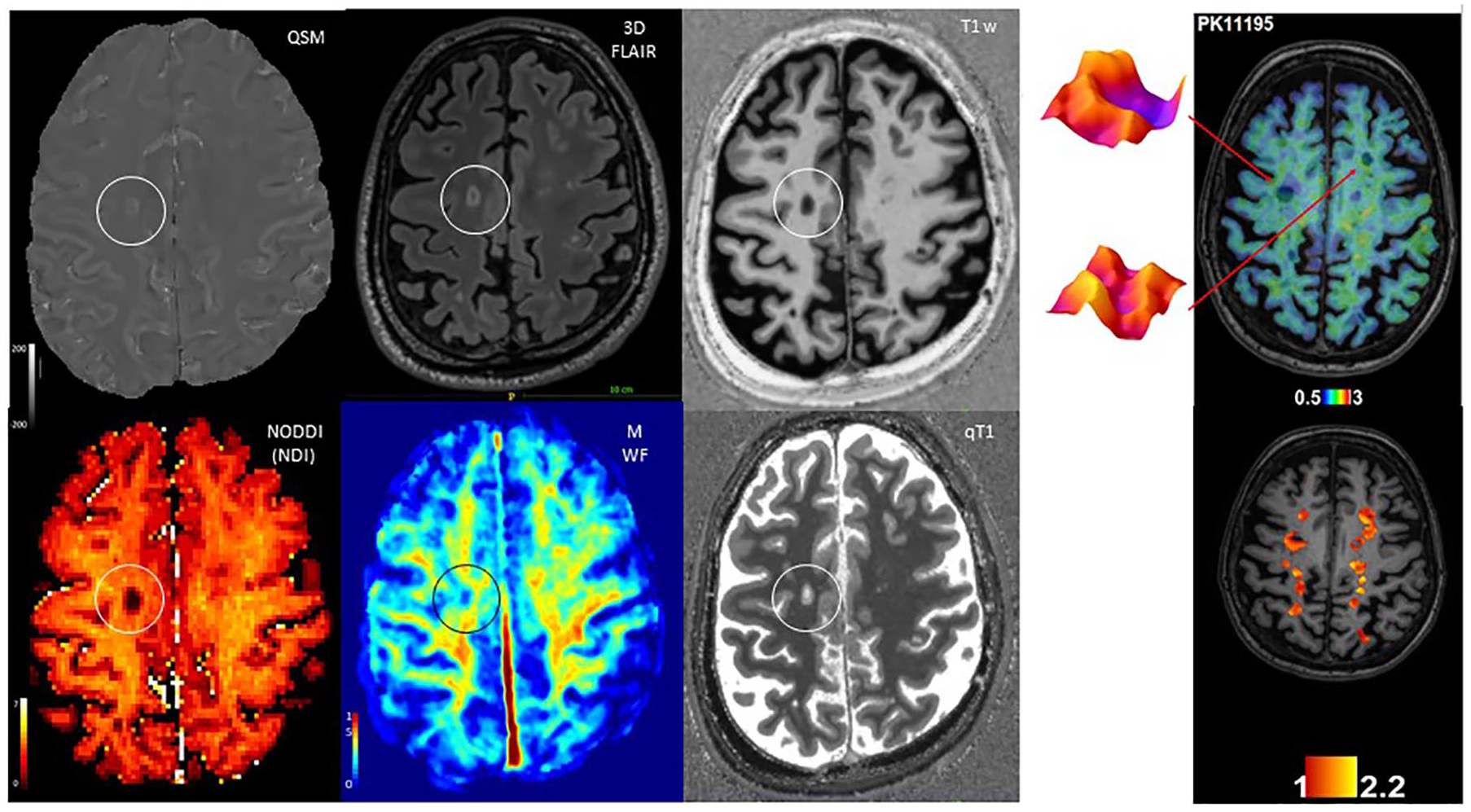
Advanced MRI and PET imaging in smouldering MS. Exemplary paramagnetic hypointense rim lesion as shown by different advanced MRI maps: Quantitative Susceptibility Mapping (QSM) shows the characteristic paramagnetic rim, while 3D FLAIR and T1w MP2RAGE images show the characteristics of a destructive lesion. The Neurite Density Index (NDI) derived from the neurite orientation dispersion and density imaging (NODDI) model, along with the myelin water fraction (MWF) and qT1 image evidence a strong reduction in axonal density, myelin and overall microstructure, respectively. The image on the right visualises comprehensive TSPO-PET measurable microglial activity in focal lesional and perilesional areas in the brain of a 48-year-old female RRMS patient with an EDSS of 4.0 and disease duration of 12 years.
With the use of double inversion recovery (DIR) sequences, diffusion tensor imaging (DTI), T1w and T2*w relaxometry, as well as magnetization transfer (MT) imaging, it has been possible to better characterise the presence of diffuse, focal and global damage in both the cortical grey and white matter, 110 which contributes to the motor and cognitive dysfunction in patients. 111 As described above, PET imaging with radiotracers can be used to target TSPO, the expression of which is associated with the density of activated innate immune cells in MS. This technique demonstrated the presence of activated macrophages and microglia in the NAWM.112,113 Interestingly, PET studies also unveiled critical roles played by the diffuse activation of innate immune cells in the slow process of tissue deterioration. Compared with RRMS, increased activation of macrophages and microglia in the NAWM in SPMS was shown to be significantly associated with disability accumulation over time51,114 and with the rate of brain atrophy. 115 It was also shown to be spatially related to microstructural damage.115,116
SELs exhibit a paramagnetic rim that is detectable in vivo by phase-contrast and susceptibility-weighted MRI.47,117 These lesions are characterised by chronic axonal loss and ongoing demyelination 118 (Figure 4). In contrast, chronically inactive MS plaques usually shrink in size on T2w and T1w MRI over the course of several years. SELs with iron rims evolve independently of acute blood-brain barrier breakdown, 118 have a lower MTR, increased radial diffusion, 118 are less likely to remyelinate, and seem to increase the vulnerability of residual axons to undergo subsequent neurodegeneration. 47 Inflammatory mediators in the hypointense lesion rim, such as free radicals or nitric oxide, may further stimulate a vicious cycle of detrimental smouldering inflammation and worsening neurodegeneration, 119 potentially accounting for the chronic disease progression in MS. 50
At postmortem, SELs were only found in the brains of patients who had reached the progressive stage of MS, 24 whereas in vivo, SELs can already be identified in patients in the relapsing stage.47,117 This suggests that the presence of SELs increases the risk of transition to progressive MS.
The ultimate consequence of both focal and diffuse, and acute and smouldering activity is the loss of brain and spinal cord volume, including specific brain structures, which relate to both motor and cognitive function and account for disability accrual. Numerous studies have shown that changes in brain volume are predictive of disease progression across all stages of MS. 120 In addition, higher rates of cervical spinal cord area loss have been associated with disability accumulation, independently of focal lesions. 121 The proportion of smouldering inflammatory activity that contributes to brain and spinal cord atrophy is unclear; however, the fact that drugs targeting acute inflammatory activity do not stop the development of progressive brain and spinal cord volume loss30,122 suggests that the contribution of smouldering processes to CNS volume loss is relevant.
Future directions
Beyond relapses and EDSS – the case for neurological stress tests
Detecting smouldering MS in clinical practice can be challenging. The EDSS is the most commonly used clinical tool, but it is not sufficiently sensitive to detect subtle changes in disability. 123 Moreover, ongoing relapses with incomplete recovery can make it difficult to pinpoint the clinical occurrence of progressive accumulation of disability. In addition, the conventional neurological examination is not adequate to monitor patients’ deterioration in their daily activities. By using an engineered glove to measure the fine motor performance of the fingers, it was demonstrated that subtle deterioration can occur even in patients with presymptomatic MS or radiologically isolated syndrome. 124 In line with these observations, the pooled analysis of the OPERA trials demonstrated that an impressively large proportion of patients, in both the ocrelizumab- and interferon-beta-1a-treated groups (87% and 78%, respectively), experienced PIRA (see Figure 1) despite being relatively early in the disease course (mean duration approximately 6 years). This was highlighted by using a composite score, which included changes in the timed 25-foot walk (T25FW), the 9-hole peg test (9HPT), or in the EDSS. 1 This approach allowed investigators to characterise elements of disease progression more comprehensively, which would most likely have been missed on routine neurological examination. Overall, a more thorough assessment of these subtle physical or cognitive changes during normal activity or under stress might help to better demonstrate the clinical effects of smouldering MS. Similarly, exercise and objective gait analysis can be used to detect subtle gait abnormalities and exercise-induced deterioration in mobility. 10 Steps are being taken to better capture these subtle changes, examples of which include the use of EDSS-plus, which is more sensitive to detect worsening in patients with SPMS, 125 and of the Overall Response Score (ORS), which is used as a primary outcome measure in some contemporary clinical trials investigating putative remyelination therapies (NCT03222973). Smart devices, such as wearables or suites of smartphone applications, can also be used to assess daily functioning. 126 The latter example involves the self-administration of different tasks to assess the impact of MS across multiple functional domains.
Activity trackers and other digital techniques such as ambient measurement systems are a feasible and popular technology for self-monitoring in MS; 127 however, the widespread use of devices is hindered by the heterogeneity of measuring techniques and algorithms, privacy concerns, and the need for adaptation specifically to MS. 128 Patient-reported outcomes are validated techniques that quantify MS patients’ own experiences of their disease, addressing several domains (depression, anxiety, fatigue, pain interference, physical function, cognition) and can be easily administered digitally via apps and software packages. 129
Changes in cognitive performance can also reflect smouldering processes in MS to some extent. The evolution of cognitive deterioration, which can occur from a very early stage of the disease, 130 is weakly associated with the inflammatory disease activity as measured by the accrual of new/enlarging T2w lesions.131,132 By using functional MRI or DTI, it has been demonstrated that cognitive impairment is better accounted for by the subtle worsening of grey matter pathology, and by network disruption and axonal degeneration, irrespective of inflammatory changes. 133
Brain health
There is mounting evidence that many additional factors that impact brain function and brain health can potentially exacerbate and interact with smouldering MS, resulting in more rapid disease worsening. These include lifestyle factors such as lack of exercise, 134 poor diet, 135 smoking 136 and excessive alcohol intake, 137 social determinants of health, 138 concomitant medications, in particular the anticholinergic burden, 139 poor sleep 140 and comorbidities. 141 Therefore, a holistic approach is needed to tackle smouldering processes and to manage MS more effectively (see Figure 5).
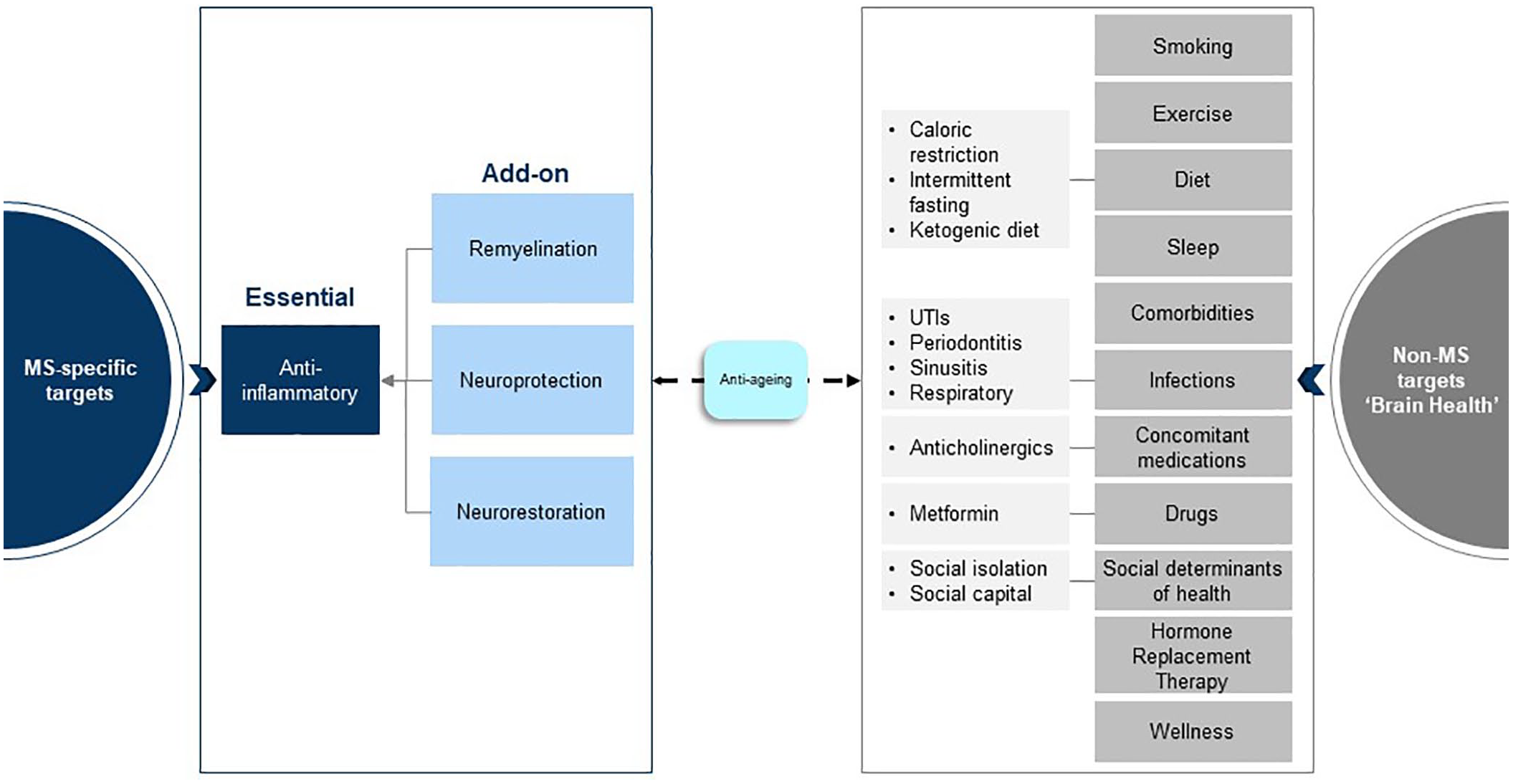
Combination therapy trials and the holistic management of MS. In addition to effective anti-inflammatory therapies, add-on neuroprotective treatments to prevent further loss of damaged or vulnerable axons and remyelination and neurorestorative therapies are required to address the pathological drivers of smouldering MS. In parallel, a holistic approach to the management of MS is required to address factors that can affect brain health and potentially accelerate MS-related end-organ damage.
Combination therapies and alternative strategies
It is evident that anti-inflammatory DMTs are insufficient to treat and manage smouldering MS. This implies that the MS community will need to turn to DMTs with dual modes of action or undertake combination therapy trials (see Figure 5). There is a compelling case for performing add-on trials of agents that target the processes discussed above, including neuroprotective agents and therapies that augment remyelination and potentially promote neurorestoration. How combination trials are designed and performed will be a challenge for regulators and the wider MS community. For example, designing studies where the primary outcome is the recovery of function is proving to be a challenge with currently available outcome measures. Another aspect to be considered is add-on neurorehabilitation to help augment recovery mechanisms by stimulating biological processes such as neuroplasticity. 142
Conclusion
We have presented evidence supporting the notion that MS as a disease entity is not focal inflammation, that is, relapses and focal MRI activity, but a more diffuse smouldering pathological process that affects the entire CNS. We therefore need to go beyond no evident inflammatory disease activity (NEIDA) and focus on other pathological processes in the end-organ (brain and spinal cord) in order to delay or prevent the slow loss of neurological function in people with MS. In addition, a holistic approach to the management of MS is needed by targeting other processes that have been shown to impact on brain health.
Footnotes
Author contributions
Disclosure
The concepts and contents of this paper emerged from several meetings hosted by Sanofi Genzyme over the preceding 2 years. Sanofi Genzyme kindly provided financial support to help adapt and design the figures and for a third party to help coordinate the submission of the manuscript. At no stage did any staff at Sanofi Genzyme have input into the writing and editing of the manuscript.
Conflict of interest statement
In the last 5 years, Gavin Giovannoni has received compensation for serving as a consultant or speaker for or has received research support from AbbVie, Aslan, Atara Bio, Biogen, BMS-Celgene, GlaxoSmithKline, GW Pharma, Janssens/Actelion, Japanese Tobacco, Jazz Pharmaceuticals, LifNano, Merck & Co, Merck KGaA/EMD Serono, Novartis, Sanofi-Genzyme, Roche/Genentech and Teva. Sanofi Genzyme kindly provided financial support to help adapt and design the figures and for a third party to help coordinate the submission of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Sanofi Genzyme kindly provided financial support to help adapt and design the figures and for a third party to help coordinate the submission of the manuscript.