
Abstract
Gastric cancer (GC) ranks among the most prevalent malignancies of the gastrointestinal system. For patients with advanced-stage disease undergoing surgical resection, postoperative 5-year survival rates remain critically low, typically below 20%. Contemporary therapeutic strategies encompass surgical intervention, cytotoxic chemotherapy, immune checkpoint blockade, and molecularly-targeted agents. Targeted therapeutics exert their effects by exploiting molecular vulnerabilities such as human epidermal growth factor receptor-2, Claudin 18.2, vascular endothelial growth factor/vascular endothelial growth factor receptor, fibroblast growth factor receptor 2, and N6-methyladenosine modification pathways, thereby suppressing tumor proliferation and offering improved clinical outcomes for advanced GC. These agents demonstrate significant potential in extending overall survival durations, underscoring their translational value and substantial research implications. This comprehensive review delineates recent advancements in GC-targeted therapies to inform precision oncology paradigms and guide future drug discovery initiatives.
Introduction
Globally, gastric cancer (GC) represents a significant oncologic burden within the gastrointestinal tract, ranking fifth in both global incidence and cancer-related mortality, with the fourth highest occurrence specifically among male patients as of 2024. 1 Epidemiologically, Chinese populations constitute approximately 37% of all GC cases worldwide.1,2 While early-stage disease remains potentially curable through surgical resection combined with cytotoxic regimens, the majority of patients present with advanced pathology upon initial diagnosis due to insidious symptom onset and delayed endoscopic evaluation. 3 Consequently, individuals with inoperable advanced GC historically faced poor prognoses owing to limited therapeutic alternatives. The recent emergence of molecularly targeted agents has transformed the therapeutic landscape for advanced-stage GC management. This review summarizes the main targets of targeted therapy in GC and its related drugs, mainly including the classic target human epidermal growth factor receptor-2 (HER2), the emerging target Claudin 18.2 (CLDN18.2), the angiogenesis target vascular endothelial growth factor (VEGF)/vascular endothelial growth factor receptor (VEGFR), and other targets under study fibroblast growth factor receptor 2 (FGFR2), N6-methyladenosine (m6A), etc., and different drug types and clinical applications for each target. The aim is to introduce the proven effective targeted therapy to medical personnel and further guide the future research direction.
This review comprehensively synthesizes recent advancements in GC-targeted therapies through a tripartite framework: mechanistically, it delineates the molecular pathways and signaling cascades underlying each therapeutic target (HER2, CLDN18.2, VEGF/VEGFR, FGFR2, m6A), providing the biological rationale for targeted intervention; clinically, it systematically evaluates completed and ongoing trials, summarizing efficacy outcomes, safety profiles, and regulatory approvals to inform evidence-based practice; and translationally, it contextualizes these findings within the evolving paradigm of precision oncology, addressing critical issues such as biomarker-driven patient selection, mechanisms of resistance, integration with immunotherapy, and the distinction between prognostic and predictive biomarkers. By integrating mechanistic insights with clinical evidence and future-oriented perspectives, this review aims to equip clinicians, investigators, and drug developers with a comprehensive understanding of the current landscape and emerging directions in GC-targeted therapy.
Throughout this review, it is important to distinguish between two fundamental roles of biomarkers in oncology. Prognostic biomarkers are associated with clinical outcomes (e.g., overall survival (OS), recurrence risk) independent of therapeutic intervention, providing information about the natural history of the disease and helping stratify patients by baseline risk. In contrast, predictive biomarkers are associated with differential response to a specific therapeutic intervention, identifying patients who are more likely to benefit from a particular treatment. A biomarker may serve one or both roles; for example, HER2 status in GC functions both as a prognostic indicator of tumor aggressiveness and as a predictive biomarker for response to HER2-directed therapies. This distinction has critical implications for clinical trial interpretation and therapeutic decision-making, as prognostic associations inform risk stratification and surveillance strategies, while predictive associations guide treatment selection. Throughout the following sections, we will explicitly clarify whether discussed biomarkers are primarily prognostic, predictive, or both.
Human epidermal growth factor receptor-2
Signal pathways and mechanisms
The clinical application of HER2-targeted therapy represents a paradigm shift in genomic medicine. Large-scale genomic analyses have confirmed that HER2 gene amplification is one of the driver alterations in GC. This genomic variation leads to increased copy numbers in the chromosomal region 17q12, resulting in HER2 protein overexpression and sustained signal activation. 4 The companion diagnostics (FISH/IHC) established based on this genomic feature enable precision patient stratification, making anti-HER2 therapy the first FDA-approved genomically guided treatment for GC. 5 HER2 (ErbB2/Neu), a member of the epidermal growth factor receptor (EGFR) family encoded at chromosome 17q12, shares structural homology with HER1, HER3, and HER4. These transmembrane receptors feature conserved molecular architecture comprising: (1) an extracellular ligand-binding domain, (2) a transmembrane segment, and (3) an intracellular tyrosine kinase (TK) region. 6 In gastric adenocarcinoma populations, HER2 expression demonstrates a spectrum spanning 12%–27% globally, with protein overexpression documented in 9%–23% of cases. 7 Chinese cohort analyses reveal concordant findings: a retrospective surgical series (n = 726) reported 13% HER2 positivity, 8 while a multicenter assessment (n = 734) identified 9.7% positivity. 9 Prevalence exhibits significant clinicopathological variation, demonstrating male predominance, intestinal-type preponderance, 10 and higher frequency in moderately versus poorly differentiated neoplasms. 11
HER2-mediated signal transduction initiates through homodimerization or heterodimerization, triggering downstream phosphorylation cascades. Dimerized HER2 directly engages the PI3K regulatory subunit p85, thereby activating the PI3K/AKT/mTOR oncogenic axis. This pathway drives GC cell proliferation, survival, and metastatic dissemination via angiogenesis induction. Concurrently, HER2 activates the MAPK/ERK signaling cascade, accelerating GC cell differentiation and migration. 12 Beyond canonical pathways, HER2 interacts with diverse molecular partners. Pathological HER2 overexpression induces mutational alterations in E-cadherin (CDH1) and RHOA GTPase, compromising intercellular adhesion complexes and augmenting metastatic potential. 13 Furthermore, HER2-amplified tumors exhibit increased CD8+ T-cell infiltration within the tumor microenvironment, paradoxically facilitating immune evasion mechanisms. 14
HER2-positive status in GC correlates with elevated postoperative recurrence risk, 7 yet its prognostic significance remains contested. 11 Contemporary evidence demonstrates divergent outcomes: prospective data associate HER2 amplification with reduced survival duration, 15 while a Japanese multicenter analysis identified HER2 overexpression as an independent predictor of mortality (hazard ratio (HR) = 1.59). 16 Contrastingly, a 2016 Chinese cohort study (n > 1500) found no survival correlation. 17 Emerging consensus suggests prognostic impact is tumor stage-dependent, with stage II patients exhibiting significantly diminished 5-year OS in HER2-positive cases. 18 Anti-HER2 antibodies mediate therapeutic effects through domain-specific binding, disrupting receptor dimerization or downstream signal transduction. The extracellular regions II and IV represent predominant pharmacologic targets. Domain IV engagement characterizes monoclonal antibodies, including Trastuzumab, its Fc-optimized derivative Margetuximab, and the antibody-drug conjugate Trastuzumab deruxtecan (T-DXd). Conversely, Pertuzumab selectively binds domain II. Bispecific constructs like Zanidatamab and KN026 achieve dual epitope recognition by simultaneously interacting with domains II and IV. 7 Beyond extracellular targeting, intracellular tyrosine kinase domain (TKD) inhibition represents an alternative strategy via tyrosine kinase inhibitors (TKIs) 19 such as Lapatinib, Tucatinib, and Pyrotinib (Figure 1).

HER2 domains and drug targets for targeted therapy in gastric cancer.
Importantly, HER2 expression is not uniform within tumors; intratumoral heterogeneity—characterized by variable HER2 copy numbers and protein levels across different tumor regions—is frequently observed in GC, with reported incidence ranging from 20% to 50% depending on detection methods and thresholds. This heterogeneity manifests both spatially (different areas within the same primary tumor or between primary and metastatic sites) and temporally (changes in HER2 status during disease progression or under treatment pressure). Such dynamic evolution poses significant challenges for accurate molecular classification, as single biopsy samples may underestimate or completely miss HER2 positivity due to sampling error, particularly in tumors with heterogeneous amplification patterns. The phenomenon of HER2 discordance between primary tumors and metastatic lesions further complicates treatment decisions, with studies reporting discordance rates of 10%–15% in GC. Under therapeutic pressure from anti-HER2 agents, HER2-negative subclones can undergo clonal selection and outgrowth, representing a key mechanism of acquired resistance. Spatial and temporal heterogeneity thus constitutes a fundamental biological determinant influencing response durability to HER2-targeted therapies, and should be systematically considered in clinical trial design, biomarker development, and patient management strategies.
It is essential to distinguish between HER2’s prognostic and predictive roles. The observed associations between HER2 positivity and postoperative recurrence risk 7 or reduced survival15,16 represent prognostic information—they reflect the natural history of HER2-driven tumors in the absence of targeted intervention or despite standard chemotherapy. However, the clinical utility of HER2 testing primarily derives from its predictive value: HER2 positivity identifies patients who derive substantial benefit from HER2-directed therapies such as trastuzumab, T-DXd, and other anti-HER2 agents. Indeed, the ToGA trial 5 and subsequent studies demonstrate that HER2-positive patients receiving targeted therapy achieve superior outcomes compared to HER2-negative populations or HER2-positive patients receiving chemotherapy alone. This dual role—adverse prognosis without targeted intervention, but excellent response to HER2 blockade—exemplifies the paradigm of “oncogene addiction” and underscores the critical importance of routine HER2 testing to identify patients who may benefit from available targeted agents. The prognostic impact of HER2 may also be stage-dependent, 16 but its predictive value remains consistent across disease stages, supporting therapeutic recommendations irrespective of prognostic considerations.
Antibody therapy
Trastuzumab mediates therapeutic effects by binding HER2 extracellular domain IV, thereby suppressing downstream oncogenic signaling. 20 Its clinical validation emerged from the pivotal 2010 ToGA phase III trial, which demonstrated significantly prolonged median OS in HER2-positive patients receiving trastuzumab-chemotherapy combination (13.8 months) versus chemotherapy alone (11.1 months). 5 This landmark evidence established Trastuzumab as the first FDA-approved anti-HER2 targeted therapy for inoperable or metastatic GC. Subsequent guidelines consistently endorse this agent: NCCN iterations (2016–2022) designate trastuzumab-based chemotherapy as first-line standard for HER2-overexpressing GC,11,21 while CSCO 2019 recommendations classify platinum/fluoropyrimidine-trastuzumab regimens as Category 1A therapy for advanced HER2-positive gastric adenocarcinoma. 22
Expanding beyond initial indications, multiple phase II trials have consistently substantiated trastuzumab’s utility in conversion and perioperative settings across various chemotherapy backbones (cisplatin, oxaliplatin/capecitabine, S-1, etc.). These studies have reported median PFS ranging from 5.9 to 9.2 months and median OS from 13.8 to 27.6 months (Table 1),23–27 collectively affirming trastuzumab’s expanding therapeutic role and the feasibility of combining it with different platinum/fluoropyrimidine regimens.
Clinical trials of trastuzumab targeted therapy in advanced gastric cancer patients.

HER2, human epidermal growth factor receptor-2; NA, not applicable.
The therapeutic utility of Trastuzumab extends substantively to perioperative management. Japan’s JACCRO GC-08 trial (2011–2012) demonstrated the efficacy of paclitaxel plus trastuzumab as second-line therapy for HER2+ advanced GC, achieving median PFS 5.1 and OS 17.1 months. The regimen was well-tolerated. 28 Concurrently, a Chinese multicenter prospective cohort study observed that in patients with non-R0 resection, the group receiving chemotherapy plus trastuzumab showed prolonged median PFS (3.1 vs 2.0 months) and OS (10.5 vs 6.2 months) compared with the control group, suggesting its potential value as an adjuvant therapy. 29 Further evidence from perioperative settings has reinforced these benefits. A 2021 Spanish phase II trial reported an exceptional median OS of 79.9 months with Trastuzumab-XELOX 30 ; two additional studies that year confirmed that neoadjuvant/adjuvant trastuzumab-chemotherapy combinations enhance pathological complete response rates and tumor regression.31,32 A 2024 French retrospective analysis further affirmed sustained survival benefits, with 80.4% 18-month DFS and 89.0% 2-year OS (Table 1). 33
Ongoing clinical investigations are actively evaluating Trastuzumab-based combination therapies across six phase II trials, incorporating diverse chemotherapeutic agents, targeted molecules, and immune checkpoint inhibitors (ICIs). The advent of PD-1-directed ICIs has revolutionized advanced GC management, establishing these agents as first-line standards that meaningfully extend long-term survival.34,35 Current research initiatives include: Sun Yat-sen University Cancer Center’s randomized phase II trial (ChiCTR2400081665) assessing SOX chemotherapy combined with Trastuzumab and PD-1 blockade in locally advanced HER2+ gastric/gastroesophageal junction (G/GEJ) adenocarcinoma; three single-arm studies examining trastuzumab-chemotherapy-ICIs combinations (ChiCTR2000039886, NCT05311189, NCT05218148); a multicenter phase II trial (NCT05997524) evaluating pCR enhancement via Trastuzumab-SOX regimens; and a Chinese phase Ib/II single-center investigation (ChiCTR2300074767) exploring Trastuzumab-XELOX-VEGFR inhibition effects on PFS in metastatic HER2+ G/GEJ cancer (Table 2). These concerted efforts herald Trastuzumab’s expanding role within precision oncology frameworks for advanced GC.
The currently ongoing clinical trial of trastuzumab for targeted therapy in patients with advanced gastric cancer.
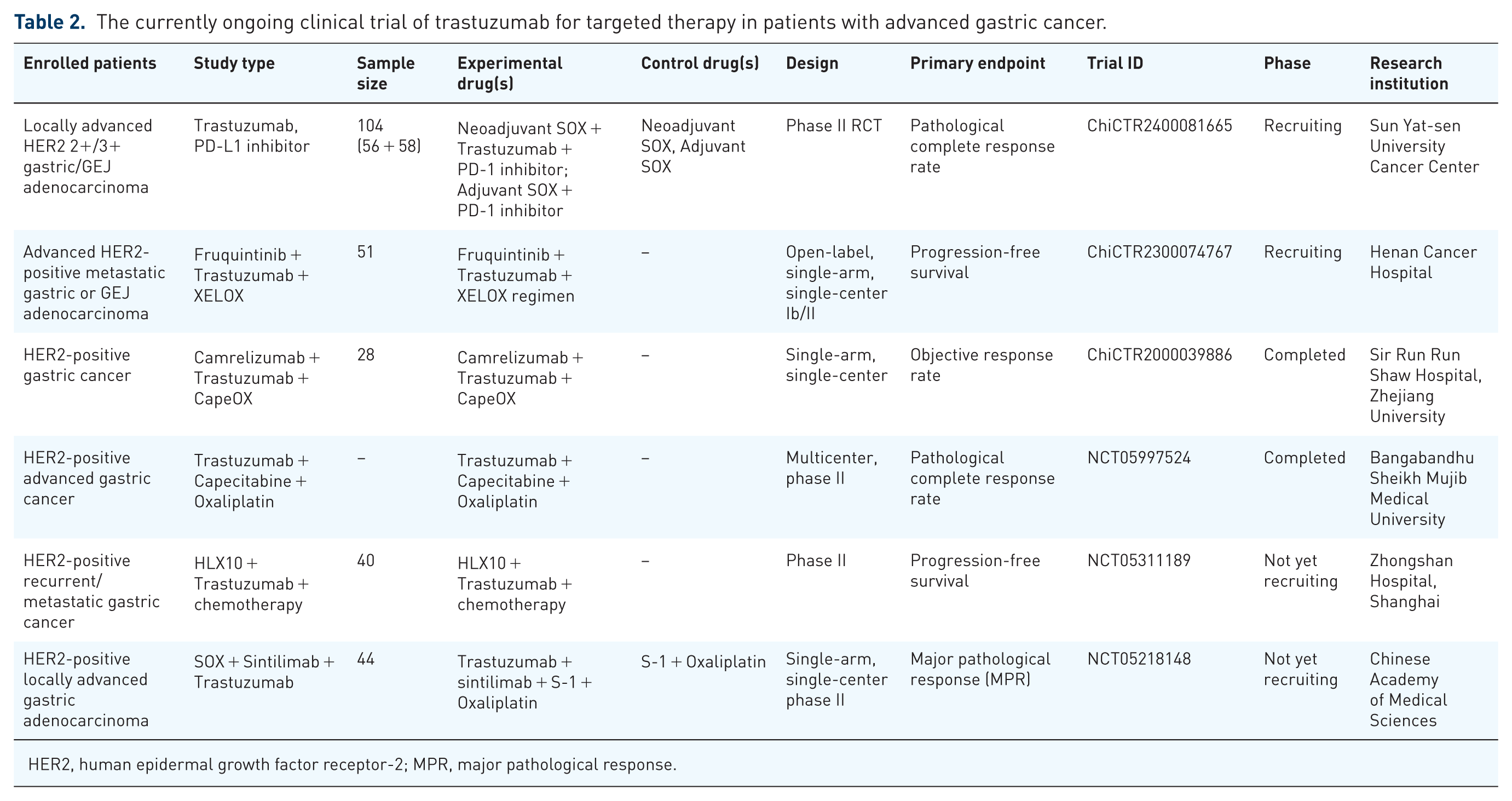
HER2, human epidermal growth factor receptor-2; MPR, major pathological response.
Beyond additive effects, emerging preclinical and clinical evidence suggests mechanistic synergy between HER2 blockade and immune checkpoint inhibition. HER2-targeted antibodies, particularly those with optimized Fc domains such as Margetuximab, can induce immunogenic cell death and enhance tumor antigen presentation, while simultaneously promoting antibody-dependent cellular cytotoxicity (ADCC) that activates innate immune effectors. This immune activation creates a favorable microenvironment for T-cell priming and infiltration, potentially converting immunologically “cold” tumors into “hot” tumors responsive to PD-1/PD-L1 blockade. Furthermore, HER2 signaling itself has been implicated in modulating the tumor immune microenvironment; HER2-amplified GC exhibit distinct patterns of immune cell infiltration, including increased CD8+ T-cell densities but also upregulation of immune checkpoint molecules as a compensatory immune evasion mechanism. The combination of HER2 inhibition with PD-1 blockade, therefore, addresses two complementary arms of anticancer immunity: direct tumor cell killing and reactivation of exhausted T-cells. Early-phase clinical trials evaluating such combinations have reported encouraging response rates, and ongoing phase III investigations (e.g., KEYNOTE-811) are expected to definitively establish whether dual HER2/PD-1 blockade can improve outcomes beyond chemotherapy plus trastuzumab alone. This paradigm exemplifies how targeted therapy can serve not only as a direct tumor-suppressive strategy but also as an immunomodulatory primer for subsequent immunotherapy efficacy.
Margetuximab shares Trastuzumab’s binding epitope but incorporates Fc domain engineering to enhance innate immune activation. This optimization augments antitumor efficacy through three mechanisms: (1) increased stimulatory Fcγ receptor engagement, (2) reduced inhibitory FcγR binding, and (3) amplified ADCC against tumor targets. 36 The 2020 CP-MGAH22-05 phase II trial established margetuximab’s favorable safety profile and clinical activity. 37 The MAHOGANY cohort A study demonstrated a favorable toxicity profile versus historical chemotherapy-trastuzumab controls using the chemotherapy-free combination of margetuximab and retifanlimab, achieving a 53% confirmed objective response rate (ORR). 38 These findings position Margetuximab as both a Trastuzumab alternative (particularly post-resistance) and a potential chemotherapy-sparing agent, thereby reducing treatment-related morbidity. This Fc optimization strategy represents a translatable platform for enhancing therapeutic antibody efficacy across oncology.
Pertuzumab represents a distinct therapeutic approach among HER2-targeting monoclonal antibodies, specifically binding the extracellular domain II rather than domain IV. Its mechanism involves sterically hindering HER2 dimerization with other EGFR family members, thereby suppressing downstream signaling cascades. 39 Initial assessment of Pertuzumab’s clinical efficacy proved contentious following the 2018 JACOB phase III trial, which demonstrated no significant OS improvement when adding Pertuzumab to Trastuzumab/chemotherapy versus placebo in metastatic HER2-positive GEJ cancer. 40 However, subsequent subgroup analyses revealed substantial geographical variation: the 2019 Chinese cohort analysis showed median DFS extending to 10.5 versus 8.6 months in controls and OS to 18.7 versus 16.1 months, 41 while Japanese data concurrently demonstrated 12.4-month DFS versus 6.3 months and 22-month OS versus 15.6 months. 42 A pivotal 2023 reanalysis of the complete JACOB dataset overturned initial conclusions, revealing statistically significant OS and PFS improvements of 3.9 and 1.3 months, respectively, thereby confirming Pertuzumab’s therapeutic value and safety. 43 Contemporary U.S. research suggests these benefits may correlate with HER2 high-copy-number alterations. 44 Current clinical evaluation through the multicenter phase II trial NCT06123338 is examining neoadjuvant Pembrolizumab-Trastuzumab-chemotherapy in resectable HER2+ GEJ tumors, potentially providing further validation of Pertuzumab’s efficacy. The modest survival gains observed in the JACOB trial—1.3 months improvement in PFS and 3.9 months in OS after reanalysis 43 —exemplify the challenge of interpreting marginal statistical benefits in advanced GC. From a statistical perspective, the HR for OS was 0.84 (95% confidence interval (CI) 0.71–1.00), indicating a 16% relative risk reduction that achieved borderline significance only after comprehensive reanalysis. The absolute benefit of 3.9 months must be contextualized against the toxicity trade-offs: the addition of pertuzumab increased grade ⩾3 adverse event rates from 64% to 72%, primarily driven by diarrhea and neutropenia. 40 Quality-of-life assessments, though not systematically reported in JACOB, are critical for determining whether the modest survival extension justifies the added treatment burden. In a disease where median survival rarely exceeds 12–15 months, even incremental improvements can be clinically meaningful, particularly if they delay symptomatic progression or enable patients to achieve important milestones. However, the number needed to treat (NNT) for one additional patient to survive at 2 years—approximately 20 based on the absolute survival difference—suggests that many patients are exposed to potential toxicity without deriving survival benefit. This underscores the importance of biomarker refinement to identify the subset of patients most likely to achieve substantial benefit, such as those with high HER2 copy number alterations. 44 For clinical decision-making, these modest benefits should be discussed with patients in the context of their individual preferences, performance status, and tolerance for potential adverse effects.
Beyond monoclonal antibodies, bispecific antibodies (BsAbs) enable multi-epitope targeting of HER2. Contemporary BsAbs predominantly inhibit HER2 signaling through simultaneous binding to extracellular domains II and IV—mechanistically analogous to combined Trastuzumab and Pertuzumab action, but with enhanced signaling blockade. ZW25 exemplifies this approach by inducing HER2 clustering through transverse domain engagement, thereby activating cellular immunity. Initial 2018 single-agent data established ZW25’s tolerability and antitumor activity in treatment-refractory HER2-positive cancers, 45 with subsequent 2021 combination studies demonstrating 68.2% ORR and 90.9% disease control rates (DCR) alongside chemotherapy. 46 Recent 2023 preclinical research further confirmed its efficacy in high-HER2 GC models. 47 The current large-scale investigation (NCT05152147, n = 918) is evaluating ZW25 with chemotherapy and tislelizumab in advanced/metastatic HER2+ GEJ malignancies.
Parallel development features KN026, which shares ZW25’s target domains. Phase II trials consistently validate its clinical utility: a 2021 study reported 55.6% ORR and 72.2% DCR after 20 weeks in frontline-refractory HER2-overexpressing G/GEJ adenocarcinoma, 48 while 2022 multicenter data confirmed 56% ORR. 49 These bispecific platforms represent promising vectors for future therapeutic innovation, including exploration of novel epitope combinations beyond domains II/IV and Fc domain optimization to enhance clinical efficacy.
The initial failure of the JACOB trial to demonstrate a significant OS benefit in the intent-to-treat population, followed by positive findings in subgroup and reanalyzed data, illustrates the critical role of patient selection and molecular heterogeneity in determining therapeutic outcomes. The geographic disparities observed—with pronounced benefits in Asian cohorts—may reflect differences in germline genetics, tumor biology, or even variations in clinical practice and subsequent therapies. More importantly, the modest survival gains of approximately 1.3 months in PFS and 3.9 months in OS, while statistically significant after reanalysis, raise the question of clinical meaningfulness. In the context of advanced GC, where prognosis remains poor, even incremental improvements can be valuable, particularly if accompanied by manageable toxicity and preserved quality of life. However, such benefits must be contextualized against the added cost and potential adverse events of dual HER2 blockade. The correlation between benefit and high HER2 copy number alterations 44 suggests that refining biomarker criteria—beyond simple HER2 positivity—could enrich for patients most likely to derive substantial benefit. Moreover, intratumoral heterogeneity in HER2 expression, where some tumor regions may harbor amplification while others do not, could explain variable responses and acquired resistance. This underscores the need for comprehensive molecular profiling, including assessment of HER2 heterogeneity and clonal evolution, to guide personalized therapeutic decisions.
Antibody-drug conjugates therapy
While antibody therapies require HER2 overexpression for efficacy-rendering them suboptimal for low-expressing HER2-positive GC 11 -antibody-drug conjugates (ADCs) overcome this limitation by delivering cytotoxic payloads to tumor microenvironments independent of target expression density. 50 T-DXd exemplifies this paradigm, comprising an anti-HER2 antibody, a cleavable linker, and a topoisomerase I inhibitor payload. A key mechanistic advantage of T-DXd and related ADCs lies in their unique “bystander effect”—the ability of the lipophilic, membrane-permeable payload to diffuse across cell membranes and kill neighboring tumor cells regardless of their HER2 expression status. This property is particularly valuable in the context of intratumoral HER2 heterogeneity, where HER2-positive and HER2-negative cell populations coexist within the same lesion. Upon binding to HER2-positive cells and undergoing internalization, the released cytotoxic payload can penetrate adjacent HER2-low or HER2-negative tumor cells, effectively eradicating heterogeneous tumor populations that would otherwise escape conventional antibody therapy. Preclinical studies have demonstrated that this bystander killing effect enables T-DXd to achieve tumor regression even in models with mixed HER2 expression patterns, providing a mechanistic rationale for its observed efficacy in HER2-low GC populations. This stands in contrast to non-cleavable linker ADCs or antibody monotherapies, which lack such diffusion capacity and remain confined to HER2-expressing cells. The bystander effect thus represents an important pharmacological strategy to overcome the therapeutic limitations imposed by tumor heterogeneity. Asian phase II trials confirmed its survival-extending potential, 50 with its unique bystander effect enabling payload diffusion to adjacent cells. Clinical validation includes: a phase I dose-escalation study demonstrating 43% ORR and 91% DCR in GEJ adenocarcinoma, notably including 16.7% low-HER2 expressors 51 ; a 2023 phase II trial reporting ORR of 26.3% (HER2 2+) and 9.5% (HER2 1+) with tumor reduction in 68.4%/60.0% of patients and median OS of 7.8/8.5 months 52 ; and corroborating phase II data showing 42% ORR across GC/G/GEJ malignancies (Table 3). 53 These findings underpinned FDA approval (January 2021) and 2023 ASCO guideline endorsement for HER2-positive G/GEJ adenocarcinoma. 54
Clinical trials of ADCs in targeted therapy for patients with advanced gastric cancer.

ADCs, antibody-drug conjugates; HER2, human epidermal growth factor receptor-2.
Ongoing clinical investigations continue to expand T-DXd’s therapeutic profile through multiple active trials. The DESTINY-Gastric03 trial (NCT04379596) exemplifies this effort as a phase Ib/II multicenter study enrolling 413 patients with advanced/metastatic HER2+ GEJ/esophageal adenocarcinoma, evaluating T-DXd-chemotherapy combinations across dose-escalation and expansion cohorts. Parallel development includes the phase III DESTINY-Gastric04 trial (NCT04704934), comparing T-DXd against ramucirumab (RAM)-paclitaxel in 490 HER2+ G/GEJ adenocarcinoma patients. Additional significant studies encompass: dual phase I/II trials assessing T-DXd-EGFR antibody combinations (NCT06085755, NCT05274048); a 257-patient prospective non-interventional analysis of T-DXd post-Trastuzumab failure in advanced HER2+ G/GEJ cancer (NCT05993234); the multicenter randomized phase II TRINITY trial examining T-DXd-SOX chemotherapy (NCT06253650); and a phase II single-arm study of T-DXd monotherapy in GC (NCT05034887; Table 4). Collectively, these investigations underscore T-DXd’s significant therapeutic potential in gastrointestinal oncology.
Ongoing clinical trials of ADCs for targeted therapy in patients with advanced gastric cancer.

ADCs, antibody-drug conjugates; GEJ, gastroesophageal junction; HER2, human epidermal growth factor receptor-2; MPR, major pathological response; NCI, National Cancer Institute; ORR, objective response rate; rwTTNT1, real-world time to next treatment. T-DXd, Trastuzumab deruxtecan.
Disitamab vedotin (RC48) represents an alternative ADC class, receiving Chinese regulatory approval in June 2021 as a second-line therapy for HER2-positive GEJ adenocarcinoma. This approval was supported by phase II single-arm trial data demonstrating 24.8% ORR, 7.9-month median OS, and 4.1-month DFS in G/GEJ malignancies. The safety profile featured 36.0% serious adverse event incidence, consistent with expected ranges (32%–43%) for advanced gastric adenocarcinoma. 55 Current investigations include: an ongoing phase III randomized multicenter trial (NCT04714190) comparing RC48 against Paclitaxel/Irinotecan/Pasireotide in 351 locally advanced/metastatic HER2-overexpressing GC patients; and a recruiting phase II/III randomized study (NCT05980481) evaluating RC48 versus Trastuzumab when combined with Toripalimab plus CAPOX chemotherapy in HER2-positive locally advanced/metastatic GC.
ARX788 represents an emerging ADC candidate demonstrating promising monotherapy efficacy in HER2-positive GEJ adenocarcinoma. Clinical evaluations report 37.9% ORR, 55.2% DCR, 10.7-month median OS, and 4.1-month DFS (Table 3). 56 Current evidence remains limited, suggesting potential as a novel therapeutic strategy warranting further investigation.
Tyrosine kinase inhibitors
In malignant cells, pathways governing cellular proliferation are typically hyperactivated. This heightened signaling necessitates receptor tyrosine kinase (RTK) proteins to engage ATP, triggering intracellular cascades that drive uncontrolled cell division. 19 TKIs, administered orally as small molecules, exert their effect by binding the intracellular TKD of RTKs. Instead of activating the kinase, TKIs compete with ATP for binding, thereby suppressing downstream signaling cascades within the HER2 family and hindering cancer cell replication. 57 Concurrently, TKI binding impedes the phosphorylation of key tyrosine residues in the PI3K/AKT and MAPK pathways. These critical pathways modulate numerous oncogenic processes, including tumor cell growth, metastasis, angiogenesis, the development of drug resistance, and the evasion of apoptosis. 58 Furthermore, the low molecular weight and pronounced lipophilicity of TKIs facilitate efficient penetration across the blood-brain barrier. This property renders them particularly valuable for targeting metastases in the central nervous system, such as those originating from breast cancer. 59 The TKD is a structural feature of HER-1, HER-2, and HER-4 proteins. Lapatinib, Tucatinib, and Pyrotinib represent specific TKI agents designed to target the HER-2 protein.
The clinical utility of Lapatinib for HER2-positive gastric adenocarcinoma continues to require extensive investigation. Findings from a 2014 phase III trial suggested that combining Lapatinib with Paclitaxel offered efficacy as a second-line regimen for HER2-overexpressing GC patients. However, this combination failed to yield a statistically significant improvement in OS. 60 This limited impact on survival endpoints was corroborated by the 2016 LOGiC phase III randomized controlled trial. Adding Lapatinib to standard therapy showed no significant benefit in either OS (12.2 vs 10.5 months) or DFS (6 vs 5.4 months) compared to placebo in advanced HER2-positive disease. While the ORR increased (53% vs 39%), Lapatinib treatment was associated with heightened gastrointestinal adverse events. 61 Similarly, a 2018 randomized, double-blind controlled study observed that metastatic GC patients exhibited a better tolerance to Lapatinib combined with ECF/X chemotherapy. Nevertheless, the addition of Lapatinib provided no significant advantage in OS or PFS outcomes, indicating minimal clinical activity for the drug in this setting. 62 While Lapatinib’s therapeutic efficacy remains unconfirmed, its safety profile appears manageable. A 2019 phase II randomized controlled trial specifically assessed the safety of incorporating Lapatinib into preoperative ECX chemotherapy. Although toxicity rates increased with Lapatinib, these effects did not preclude surgical intervention. 63 Consequently, the potential role of Lapatinib in GC management warrants further rigorous evaluation.
Clinical evidence supporting Tucatinib’s efficacy in GC remains limited, with no completed controlled trials currently validating its therapeutic potential. Preclinical investigations have explored its mechanisms of action: a 2019 study engineered HER2-specific chimeric antigen receptor (CAR)-expressing NK cells (Z-cells) and demonstrated that Tucatinib co-administration enhanced Z-cell infiltration into bulky tumor xenografts, potentiating antitumor effects. 64 Additional experimental work indicates that Tucatinib monotherapy exhibits activity against HER2-positive cell lines, and combination with Trastuzumab or Docetaxel yields synergistic antitumor effects and improved DCRs. In GC-specific models, Tucatinib suppressed tumor growth in NCI-N87 xenografts, with the Tucatinib-Trastuzumab combination inducing superior regression compared to either agent alone. 65 Clinical evaluation is ongoing with the phase II/III MOUNTAINEER-02 trial (initiated 2022), investigating Tucatinib combined with Trastuzumab, RAM, and Paclitaxel as therapy for HER2-positive G/GEJ adenocarcinoma; results are awaited. 66 Further investigation into single-agent activity and the translational potential of Z-cell/Tucatinib combinations remains warranted.
Notably, the Tucatinib-Trastuzumab combination induced superior regression rates compared to monotherapies. Clinical evaluation advanced with the 2022 initiation of MOUNTAINEER-02 (phase II/III), investigating Tucatinib combined with Trastuzumab, RAM, and Paclitaxel as adjuvant therapy for HER2-positive G/GEJ adenocarcinoma, though results remain pending. Collectively, while Tucatinib’s clinical translation progresses with anticipation for MOUNTAINEER-02 outcomes, further investigation into its single-agent efficacy is warranted. The Z-cell/Tucatinib combination represents a promising translational avenue requiring comprehensive preclinical and clinical validation.
Early clinical evaluation (NCT02378389, 2014-2017) established Pyrotinib’s tolerability in HER2-positive GC patients, though therapeutic efficacy proved limited. Subsequent translational research in 2020 addressed this efficacy gap through mechanistic exploration. Utilizing human GC cell lines and AVATAR murine models, investigators delineated Pyrotinib resistance pathways and formulated a combinatorial strategy incorporating the CDK4/6 inhibitor SHR6390.
Preclinical validation in AVATAR systems preceded promising clinical observations: five patients receiving Pyrotinib-SHR6390 combination therapy achieved DFS intervals of 120, 200, 532, 109, and 57 days, 67 indicating Pyrotinib’s developmental promise. Subsequent investigations prioritized safety profiling. A 2023 phase I dose-escalation study confirmed manageable toxicity for Pyrotinib monotherapy and Pyrotinib-Docetaxel neoadjuvant regimens in HER2-positive GC. 68 That same year, multicenter phase I data demonstrated encouraging clinical activity for first-line Pyrotinib-Camrelizumab-chemotherapy in advanced HER2-positive G/GEJ adenocarcinoma (ORR with 77.8%), with adverse events aligning with projected safety parameters. 69
Notably, 2023 mechanistic research revealed Pyrotinib’s radiosensitizing effect in HER2-positive GC models. This function was mediated through suppression of radiation-induced HER2 nuclear translocation and ERK1/2 signaling pathway inhibition, 70 unveiling its adjuvant potential in radiotherapy and establishing a novel translational framework for TKIs. Despite the clinical efficacy of HER2-targeted therapies, acquired resistance inevitably develops in most patients, limiting long-term treatment benefit. Multiple resistance mechanisms have been identified in GC. Secondary HER2 alterations include loss of HER2 expression under therapeutic pressure—observed in up to 30% of post-progression biopsies—and acquired HER2 mutations within the TKD that impair drug binding. Receptor shedding, mediated by metalloproteases such as ADAM10/17, cleaves the HER2 extracellular domain, generating truncated p95-HER2 fragments that lack the trastuzumab-binding epitope but retain constitutive kinase activity, conferring resistance to antibody-based therapies while potentially preserving sensitivity to TKIs. Alternative pathway activation represents a major escape mechanism: upregulation of other RTKs (MET, EGFR, HER3) or activation of downstream signaling nodes (PI3K/AKT, RAS/MAPK) can bypass HER2 blockade. MET amplification, in particular, has been documented in trastuzumab-resistant GC models and patient samples. PI3K pathway alterations, including PIK3CA mutations or PTEN loss, sustain AKT signaling independently of upstream HER2 input. The tumor microenvironment also contributes to resistance, with stromal cells secreting growth factors that activate compensatory survival pathways. Understanding these heterogeneous resistance mechanisms has important therapeutic implications: repeat biopsy at progression may identify targetable alterations (e.g., MET amplification) that guide subsequent therapy selection; combination strategies targeting parallel pathways (e.g., HER2 + MET inhibitors) are under investigation; and agents with distinct mechanisms of action, such as T-DXd which retains activity against trastuzumab-resistant tumors due to its cytotoxic payload, provide effective salvage options.
Claudin18.2
Molecular pathways and mechanisms
Claudins (CLDNs) constitute essential structural elements of cellular tight junctions. These transmembrane proteins feature extracellular loop domains that present viable targets for diagnostic and therapeutic applications. 71 The 27-member CLDN family demonstrates tissue-specific expression patterns across diverse malignancies. 72 CLDN18.2, a 264-amino acid transmembrane protein (~27 kDa) generated through CLDN18 gene alternative splicing, features four transmembrane segments, two extracellular loops (ECL1/ECL2), and dual cytoplasmic regions. 73 Its functionally critical N-terminal domain (69 amino acids) contains eight ECL1 residues that confer gastric mucosa specificity and enable selective antibody binding (e.g., Zolbetuximab), distinguishing it from the homologous CLDN18.1 isoform.74,75 The C-terminus harbors a PDZ-binding motif that anchors to F-actin via ZO-1 scaffolding protein interaction, preserving epithelial polarity and barrier integrity. 76 Alternative splicing of exon 1 from the chromosome 3q22-located CLDN18 gene yields two isoforms: CLDN18.1 and CLDN18.2, sharing substantial sequence homology. 75 CLDN18.2 exhibits highly restricted expression, limited to differentiated gastric mucosal epithelia and absent in extragastric normal tissues.77,78 It critically maintains gastric barrier function, cellular polarization, and acid resistance by forming paracellular seals against hydrochloric acid. 79 Malignant transformation disrupts cellular architecture, exposing CLDN18.2 epitopes on tumor cell surfaces, resulting in stable and selective expression within gastric carcinoma. 74
Clinical prevalence studies report CLDN18.2 positivity in 29.4% (150/510) of primary GC and 34.1% (45/132) of metastatic lesions, correlating significantly with non-antral location, Lauren diffuse type, and EBV-associated subtypes. 80 Additional investigations confirm expression in 45% of 300 GC patients 81 and 46.5% (139/299) in stage I–III cohorts. 82 This substantially exceeds HER2 positivity rates (approximately 20%), potentially benefiting a larger GC patient population through targeted approaches (Figure 2).

Claudin18.2 domains and drug targets for targeted therapy in gastric cancer.
CLDN18.2-positive GCs demonstrate distinct biological behaviors, including reduced lymph node metastasis (55.4% vs 66.7%) and decreased lymphovascular invasion (56.1% vs 75.5%) compared to CLDN18.2-negative tumors, though perineural invasion rates remain comparable. 82 Earlier work corroborates the inverse association with perineural infiltration. 83 Prognostically, CLDN18.2 expression correlates with improved outcomes: patients exhibit superior 3-year recurrence-free survival (83.6% vs 73.7%) and OS (85.6% vs 81.3%), 82 consistent with earlier findings of prolonged OS in expressors. 83 These molecular characteristics, frequent expression, tissue specificity, and favorable prognostic associations collectively underscore CLDN18.2’s viability as a therapeutic target in systemic GC management. The observation that CLDN18.2 expression correlates with improved 3-year recurrence-free survival (83.6% vs 73.7%) and OS (85.6% vs 81.3%), 82 along with prolonged OS in expressors, 83 indicates a prognostic role for this biomarker. CLDN18.2-positive GC appear to exhibit less aggressive biological behavior, with reduced lymph node metastasis and decreased lymphovascular invasion, 82 suggesting that CLDN18.2 expression may mark a distinct tumor subtype with intrinsically better outcomes.
Antibody therapy
Zolbetuximab stands as the sole clinically validated CLDN18.2-targeted therapeutic, supported by robust trial evidence. 84 This chimeric IgG1 monoclonal antibody (mAb) demonstrates high-affinity, selective binding to CLDN18.2, inducing cell death in CLDN18.2-positive G/GEJ adenocarcinomas through ADCC and complement-mediated mechanisms. 85 Clinical validation emerged from the 2021 FAST phase II trial, where adding Zolbetuximab to first-line epirubicin + oxaliplatin + capecitabine chemotherapy significantly improved outcomes in CLDN18.2-overexpressing patients: median OS increased from 8.3 to 13 months, median PFS from 5.3 to 7.5 months, ORR from 25% to 39%, and DCR from 76.2% to 83.1%. 86 Subsequent 2023 phase III data confirmed these benefits, showing Zolbetuximab + mFOLFOX6 extended PFS (10.61 vs 8.67 months) and OS (18.23 vs 15.54 months) in CLDN18.2-positive GEJ adenocarcinoma. 87 Another phase III trial that year demonstrated consistent efficacy: Zolbetuximab + CAPOX improved median OS (14.39 vs 12.16 months) and PFS (8.21 vs 6.8 months) versus CAPOX alone. 88
Beyond combination regimens, Zolbetuximab monotherapy exhibits clinical activity. A 2019 phase II trial reported a 14% ORR in G/GEJ adenocarcinoma patients with moderate-to-high CLDN18.2 expression, 89 and a 2023 Japanese phase I study showed monotherapy achieved a median OS of 4.4 months and DCR of 64.7%. 90 In the pivotal phase III SPOTLIGHT and GLOW trials, the most common treatment-emergent adverse events associated with zolbetuximab plus chemotherapy were nausea (68.5%–82% all-grade) and vomiting (66.1%–67% all-grade), with grade ⩾3 events occurring in 8.7%–16% of patients.87,88 These events were most frequently observed during the first treatment cycle and decreased in subsequent cycles, highlighting the importance of appropriate supportive care.87,88 Based on the protocols and safety management strategies from these trials, appropriate supportive care consists of a proactive antiemetic prophylaxis regimen administered prior to each zolbetuximab infusion, including 5-HT3 receptor antagonists (e.g., ondansetron or granisetron), NK1 receptor antagonists (e.g., aprepitant), and/or corticosteroids (e.g., dexamethasone). In cases of breakthrough nausea or vomiting, dose interruptions, infusion rate adjustments, and the addition of supplemental antiemetics (e.g., metoclopramide) are recommended to improve tolerability and reduce the risk of treatment discontinuation. 88 These collective findings support Zolbetuximab’s incorporation into clinical guidelines as a standard option for CLDN18.2-positive/HER2-negative locally advanced or metastatic G/GEJ adenocarcinoma (Table 5), 88 provided that appropriate supportive care measures are implemented to manage gastrointestinal toxicities.
Zolbetuximab targeting therapy clinical trial in patients with advanced gastric cancer.
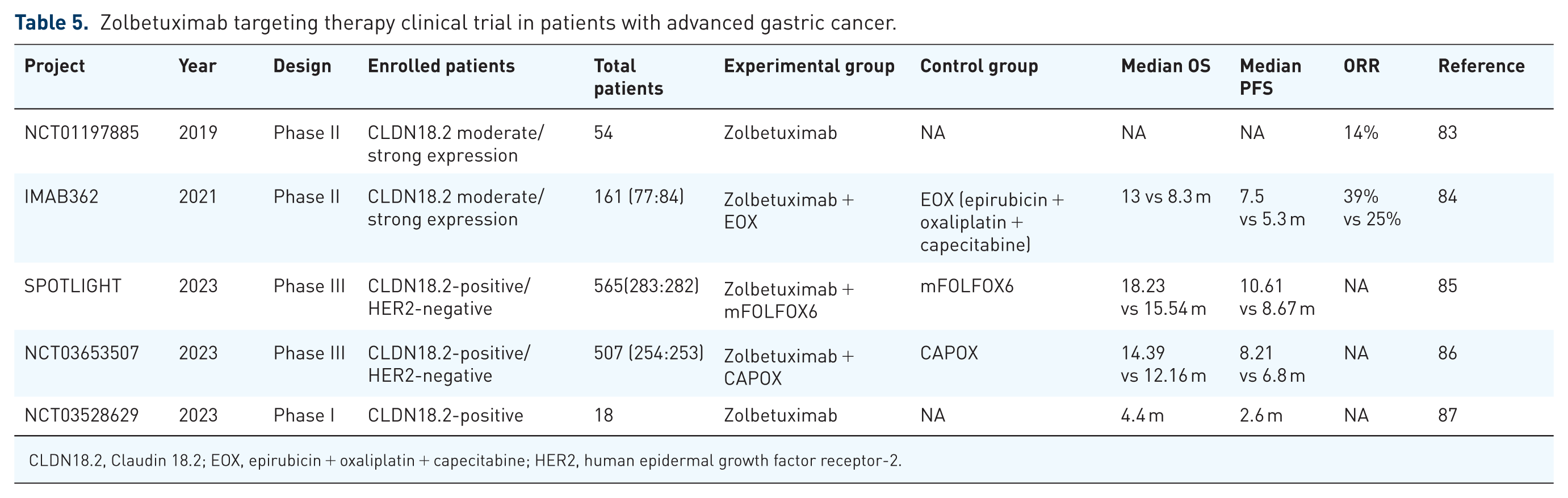
CLDN18.2, Claudin 18.2; EOX, epirubicin + oxaliplatin + capecitabine; HER2, human epidermal growth factor receptor-2.
Current investigation continues through the Japanese phase II trial NCT03505320, which is enrolling 143 patients with recurrent/metastatic CLDN18.2+ G/GEJ adenocarcinoma. This study evaluates Zolbetuximab combined with chemotherapy and/or immunotherapy, aiming to further define its monotherapy utility and combination efficacy profiles.
CLDN18.2-targeted CAR T cells (CT041)
CT041 CAR-T cells incorporate an engineered construct featuring a humanized anti-CLDN18.2 scFv, CD8α hinge, CD28 transmembrane/costimulatory domains, and CD3ζ activation domain, enabling precise targeting of CLDN18.2-positive malignancies. Initial clinical validation emerged from a 2022 phase I trial where CT041 achieved a 48.6% ORR in G/GEJ adenocarcinoma, with 81.2% 6-month survival. 91 Concurrent multicenter phase I data confirmed both safety and significant antitumor activity in chemotherapy-experienced advanced gastrointestinal cancer patients. 92 Recent 2024 mechanistic studies further demonstrated gastric adenocarcinoma cell susceptibility to CT041. 93
CAR-T therapy offers distinct advantages over conventional treatments, including potential durable remission and reduced hospitalization requirements, substantially improving quality of life metrics. However, significant challenges impede solid tumor applications: tumor heterogeneity, dense stromal architecture, and immunosuppressive microenvironments limit response durability compared to hematologic malignancies. 94 Additionally, prohibitive manufacturing costs restrict accessibility for most GC patients.
Ongoing investigation through the Chinese open-label phase Ib/II trial NCT04581473 (n = 192) aims to characterize CT041’s efficacy, safety, and pharmacokinetics in advanced G/GEJ adenocarcinoma and pancreatic cancer. Completion anticipated in 2038, this study may address critical barriers by establishing novel efficacy optimization strategies and cost-reduction methodologies.
Vascular endothelial growth factor/vascular endothelial growth factor receptor
Molecular pathways and mechanisms
Malignant progression predominantly occurs in highly vascularized tissues (e.g., pulmonary, hepatic), where tumor neovascularization significantly increases risks of lymphatic dissemination and hematogenous spread. 95 The foundational concept of tumoral angiogenesis emerged in 1971 with the hypothesis that malignancies secrete specific factors to initiate neovascular formation. 96 As expanding tumor mass outpaces local vascular support, hypoxia and nutrient deprivation trigger overexpression of VEGF—a critical mitogen sustaining uncontrolled proliferation. This dysregulation is documented across multiple epithelial malignancies, including esophageal, pulmonary, mammary, renal, and colorectal carcinomas. 97 VEGF was subsequently characterized in 1989 as a potent pro-angiogenic signaling molecule isolated from bovine pituitary folliculostellate cells. 98 Progressive refinement of preclinical models 99 facilitated the translational development of VEGF-targeted mAb therapeutics.
The VEGF signaling network comprises five principal ligands—VEGF-A, VEGF-B, VEGF-C, VEGF-D, and PlGF—that engage three class III RTKs (VEGFR-1, VEGFR-2, and VEGFR-3). 100 Ligand binding occurs through immunoglobulin-like domains within the extracellular region, triggering signal transduction via transmembrane and intracellular TK domains. 100 This molecular interaction induces receptor dimerization and subsequent TK autophosphorylation, activating three core pathways: the PLCγ-PKC-Raf-MEK-ERK cascade, the PI3K-AKT-mTOR axis, and the FAK-Src-Rac circuit.101,102 Collectively, these signaling conduits regulate enhanced vascular permeability, pathological angiogenesis, and lymphangiogenesis, thereby accelerating tumor progression and metastatic dissemination. Therapeutic interventions disrupt this paradigm through distinct mechanisms: Bevacizumab neutralizes circulating VEGF-A to inhibit aberrant vasculogenesis, 103 while RAM competitively blocks ligand binding to VEGFR2, suppressing downstream angiogenic signaling. 104 Tumors frequently develop resistance through compensatory pathway activation and vascular niche remodeling, 105 as illustrated in Figure 3.

VEGF/VEGFR domains and drug targets for targeted therapy in gastric cancer.
Ramucirumab
RAM functions as a recombinant mAb targeting VEGFR-2 with high specificity. By binding to VEGFR-2, it competitively inhibits interactions with cognate ligands VEGF-A, VEGF-C, and VEGF-D, thereby disrupting pro-angiogenic signaling cascades. 106 Initial clinical validation emerged from the pivotal 2014 REGARD phase III trial, which established RAM monotherapy’s efficacy and safety in previously treated advanced GC. 104 This foundational evidence was rapidly augmented by the landmark RAINBOW study published concurrently. This randomized, double-blind phase III investigation demonstrated significantly improved OS for RAM combined with paclitaxel versus placebo, cementing the combination’s status as standard second-line therapy for advanced gastric carcinoma. 107
RAM’s therapeutic benefit demonstrated population-specific validation through the 2021 RAINBOW-Asia subgroup analysis. This regional investigation confirmed significant extension of median PFS (4.14 vs 3.15 months) and OS (8.71 vs 7.92 months) in Asian patients receiving combination therapy. 108 Concurrent with ICIs in GC management, researchers explored RAM-immunotherapy synergy. The multicenter JVDJ Ib/II trial (2020) evaluated RAM plus PD-L1 inhibitor Durvalumab, demonstrating manageable toxicity and antitumor activity across cohorts, including G/GEJ adenocarcinoma patients. This regimen produced a marked improvement in median PFS (12.4 vs 2.6 months), with enhanced efficacy observed in PD-L1-high expressors. 109 Parallel investigations examined alternative chemotherapeutic partners, as evidenced by the 2022 HGCSG1603 phase II study. RAM combined with Irinotecan achieved a median PFS of 4.2 months and OS of 9.6 months in advanced GC, confirming both clinical efficacy and acceptable safety for this second-line combination approach. 110
A notable mechanistic insight surfaced during the ICIs era: 2020 research revealed that metastatic GEJ adenocarcinoma patient’s refractory to prior ICIs exhibited significantly enhanced ORR and prolonged PFS (12.2 vs 3.0 months) when treated sequentially with RAM (VEGFR-2 antagonist) plus Paclitaxel, compared to ICIs-naïve counterparts. This profound clinical benefit correlated with reduced regulatory T-cell (Treg) infiltration within the tumor immune niche, indicating that preceding ICIs exposure potentially reprograms the tumor microenvironment to potentiate subsequent anti-angiogenic efficacy. 111 The immunomodulatory effects of VEGF/VEGFR inhibition extend beyond the previously described reduction in Treg infiltration. Mechanistically, VEGF signaling contributes to an immunosuppressive tumor microenvironment through multiple parallel pathways: it promotes the expansion and accumulation of myeloid-derived suppressor cells, inhibits dendritic cell maturation and antigen presentation capacity, and induces endothelial cell anergy that impairs lymphocyte extravasation into tumor sites. VEGFR-2 blockade with RAM therefore reverses these immunosuppressive effects, effectively “reprogramming” the tumor immune landscape. The clinical observation that prior ICI exposure enhances subsequent RAM efficacy 111 suggests a sequential therapeutic paradigm: initial immunotherapy primes and expands tumor-specific T-cell populations, while subsequent anti-angiogenic therapy facilitates their infiltration into tumors by normalizing aberrant vasculature and removing immunosuppressive barriers. This “priming and infiltration” model has important implications for treatment sequencing strategies. Moreover, the magnitude of clinical benefit observed in ICI-refractory patients receiving RAM-with PFS extended to 12.2 months compared to 3.0 months in ICI-naïve counterparts indicates that the immunomodulatory effects of VEGFR-2 inhibition may be most pronounced in a T-cell-inflamed microenvironment. These mechanistic insights argue for rational combination and sequencing strategies that leverage the complementary biological effects of immunotherapy and anti-angiogenic agents, rather than their empirical co-administration.
The survival benefits demonstrated in the REGARD and RAINBOW trials warrant careful contextual interpretation. In REGARD, RAM monotherapy improved median OS by 1.4 months compared to placebo (5.2 vs 3.8 months; HR 0.78, 95% CI 0.60–0.99). 104 The absolute benefit of 1.4 months represents a 22% relative reduction in the risk of death, with a NNT of approximately 8 to prevent 1 death at 6 months. In RAINBOW, the addition of RAM to paclitaxel extended median OS by 2.3 months (9.6 vs 7.4 months; HR 0.81, 95% CI 0.68–0.96) and median PFS by 1.5 months (4.4 vs 2.9 months; HR 0.64, 95% CI 0.54–0.75). These benefits must be balanced against toxicity considerations: the RAM-paclitaxel combination increased rates of grade ⩾3 neutropenia (41% vs 19%), hypertension (14% vs 2%), and fatigue (12% vs 5%), though febrile neutropenia rates were similar. Quality-of-life analyses from RAINBOW demonstrated that the combination delayed deterioration in global health status and several functional scales compared to paclitaxel alone, suggesting that the survival benefit was achieved without compromising patient-reported outcomes. 107 The clinical meaningfulness of these modest survival gains is supported by the favorable risk-benefit profile and the lack of effective alternatives in the second-line setting. However, the observation that prior ICI exposure enhances subsequent RAM efficacy 111 —with PFS extended to 12.2 months in ICI-refractory patients versus 3.0 months in ICI-naïve counterparts—illustrates how patient selection based on prior treatment history can dramatically alter the absolute benefit magnitude, emphasizing the importance of treatment sequencing and personalized decision-making.
RAM’s therapeutic frontier now extends to earlier treatment lines, evidenced by a 2023 multicenter randomized phase II/III investigation. This trial assessed FLOT chemotherapy augmented with RAM (FLOT-Ram) for perioperative management of resectable GEJ adenocarcinoma. Interim analysis demonstrated superior efficacy for the combination arm, with statistically significant improvements in both median progression-free and OS relative to FLOT alone, 112 establishing its promising role within the neoadjuvant-adjuvant continuum.
Emerging evidence continues to refine RAM’s therapeutic utility within optimized treatment sequences and responsive patient cohorts. A 2024 retrospective analysis of second-line trials demonstrated that advanced GEJ cancer patients achieving complete or partial response to first-line FOLFOX plus Nivolumab/Ipilimumab dual immunotherapy derived substantial survival benefits when receiving second-line RAM-chemotherapy combinations. Compared to non-RAM regimens, these patients exhibited significantly extended OS measured from first-line initiation (28.9 vs 16.5 months), improved second-line OS (9.6 vs 7.5 months), superior PFS (5.6 vs 2.9 months), and enhanced DCR (53% vs 29%). 113 The broader cohort analysis confirmed a consistent second-line PFS advantage (4.5 vs 2.9 months), reinforcing that profound responders to frontline immunochemotherapy constitute the optimal subgroup for deriving maximal benefit from sequential RAM-based interventions.
Apatinib (Rivoceranib)
Apatinib emerged in 2022 as an investigational targeted antiangiogenic agent for gastrointestinal malignancies (not yet globally approved), demonstrating efficacy in clinical studies both as monotherapy and in combination regimens with immunotherapies. Its primary mechanism involves potent inhibition of multiple oncogenic kinases—particularly VEGFR-2—through selective small-molecule TK inhibition. This molecular targeting induces tumor cell apoptosis and significantly curbs proliferation across diverse solid tumors. 114 Significantly, Apatinib’s activity extends beyond angiogenesis suppression: it overcomes chemoresistance by resensitizing malignant cells to conventional cytotoxic agents and reverses ATP-binding cassette transporter-mediated multidrug resistance. 115 These dual pharmacological actions provide a robust mechanistic rationale for combinatorial approaches with chemotherapy.
Initial validation of Apatinib’s clinical utility emerged from monotherapy studies, notably a 2023 single-center retrospective analysis. This investigation demonstrated that patients with unresectable stage III/IV or metastatic GEJ adenocarcinoma receiving Apatinib monotherapy achieved a median PFS of 4.2 months and OS of 9.3 months. 116 These findings established preliminary efficacy benchmarks for subsequent clinical development.
The 2024 Apatinib research landscape demonstrated substantial advancement through multiple pivotal clinical reports exploring diverse therapeutic applications. In frontline settings, a phase I trial evaluating Camrelizumab-Apatinib-chemotherapy triple therapy for advanced G/GEJ adenocarcinoma demonstrated marked efficacy with 76.5% ORR and 8.4-month median PFS, 117 establishing preliminary evidence for this combination’s potent antitumor activity. For second-line metastatic GC, a phase II investigation of PD-1 inhibitor plus nab-Paclitaxel and Apatinib yielded a median OS of 10.1 months and PFS of 6.2 months, 118 confirming meaningful clinical benefit in pretreated populations. Later-line treatment strategies were revolutionized by the international ANGEL phase III trial, where Apatinib monotherapy significantly outperformed placebo in refractory advanced/metastatic G/GEJ cancer: PFS improved (2.83 vs 1.77 months), ORR increased (6.5% vs 3%), and DCR substantially elevated (40.3% vs 13.2%), 119 providing robust evidence for further investigation and potential standardization of Apatinib in salvage therapy. Perioperative management advanced through the DRAGON IV/CAP 05 phase III study, where Camrelizumab-Apatinib-SOX neoadjuvant therapy significantly elevated pCR rates versus SOX alone (18.3% vs 5.0%), 120 suggesting enhanced curative potential for locally advanced GC patients.
Others
The therapeutic landscape for gastrointestinal cancers continues to evolve beyond established anti-angiogenic agents, with extensive exploration of novel inhibitors exhibiting diverse targeting profiles and mechanisms. This expanding research frontier encompasses highly selective VEGFR antagonists, multi-targeted kinase inhibitors, and innovative immunomodulatory combination approaches, collectively advancing treatment paradigms. Fruquintinib exemplifies this progress as an oral, highly selective small-molecule inhibitor targeting VEGFR1, VEGFR2, and VEGFR3. Having demonstrated clinically meaningful activity in gastrointestinal malignancies, it has received Chinese approval for chemotherapy-refractory metastatic colorectal cancer, with ongoing phase III trials further characterizing its therapeutic potential.
BC001, a novel fully human IgG1 mAb targeting VEGFR2 for advanced solid tumors, entered clinical evaluation in 2024. Initial phase I investigations demonstrate acceptable safety profiles and preliminary antitumor activity both as monotherapy and in combinatorial approaches with conventional chemotherapy. Notably, among GC patients, BC001 demonstrated validated efficacy metrics including 28.6% ORR, 5.4-month median PFS, and 9.4-month OS. 121 These findings establish BC001 as a promising therapeutic candidate for advanced GC, necessitating expanded clinical evaluation in larger cohort studies.
Vandetanib represents an alternative therapeutic approach through its function as an oral multi-kinase inhibitor directed against VEGFR2, EGFR, and RET. Early-phase exploratory trials indicate potential for concurrent pathway blockade across these molecular targets. 122 Beyond direct kinase inhibition, innovative immunomodulatory strategies targeting VEGFR2 have emerged. A 2023 investigation demonstrated that αVEGFR2-MICA fusion antibodies substantially potentiate immunotherapy responses and exhibit synergistic activity with PD-1 blockade, 123 establishing a mechanistic foundation for developing integrated therapeutic approaches that concomitantly address angiogenesis and immune evasion.
Sorafenib, an established multi-kinase inhibitor targeting VEGFR and RAF-MEK-ERK pathways, demonstrates well-characterized antiangiogenic properties. 124 Nevertheless, its therapeutic augmentation potential in GC necessitated rigorous assessment. The randomized phase II STARGATE trial (2023) directly evaluated this by comparing Capecitabine plus Cisplatin (XP) with the XP-Sorafenib combination in metastatic GC. Findings revealed that while the Sorafenib-containing regimen exhibited manageable toxicity, it did not demonstrate superior efficacy compared to XP chemotherapy alone. 125 This outcome highlights that combinatorial approaches incorporating multi-targeted kinase inhibitors do not universally enhance conventional chemotherapy efficacy in GC, emphasizing the critical importance of molecular target refinement and biomarker-driven patient selection.
Resistance to anti-angiogenic therapy represents a major clinical challenge, with multiple compensatory mechanisms limiting the durability of VEGF/VEGFR blockade. VEGF pathway escape involves upregulation of alternative pro-angiogenic factors, including fibroblast growth factors (FGFs), platelet-derived growth factors (PDGFs), angiopoietins, and ephrins, which can sustain tumor vascularization despite VEGFR-2 inhibition. Vascular niche remodeling includes increased pericyte coverage of tumor vessels, providing structural support and survival signals that render endothelial cells less dependent on VEGF signaling. Hypoxia-driven adaptations occur as anti-angiogenic therapy exacerbates tumor hypoxia, stabilizing hypoxia-inducible factors that transcriptionally activate a broad program of pro-angiogenic and invasive genes, promoting a more aggressive tumor phenotype. Metabolic reprogramming enables tumor cells to adapt to reduced oxygen and nutrient availability, shifting toward glycolytic metabolism and enhancing survival under hypoxic stress. Immune microenvironment modulation also contributes to resistance: chronic VEGF blockade can alter immune cell composition, potentially promoting immunosuppressive populations that support tumor growth through non-angiogenic mechanisms. Importantly, these resistance mechanisms are not mutually exclusive and often co-exist within heterogeneous tumor populations. Strategies to overcome resistance include rational combination with inhibitors of compensatory pathways (e.g., FGFR inhibitors), normalization of the remaining vasculature to improve drug delivery, and sequential therapy approaches that re-induce VEGF dependence after a treatment break. The clinical observation that some patients respond to reintroduction of anti-angiogenic therapy after progression suggests that resistance may be partially reversible, warranting further investigation into optimal treatment sequencing.
The research landscape of anti-angiogenic therapy for gastrointestinal cancers is becoming increasingly diversified and refined. Future investigations will focus on: Elucidating the mechanisms of action and resistance patterns of various inhibitors; optimizing drug selection and combination strategies with immunotherapy and chemotherapy; precisely identifying patient subgroups most likely to benefit through biomarker-guided approaches; and accelerating pivotal clinical trials for promising new agents like BC001—all aimed at ultimately improving survival outcomes across the spectrum of gastrointestinal malignancies.
Fibroblast growth factor receptor
FGFRs significantly influence GC pathogenesis and modulate tumor immune microenvironments, serving as potential prognostic biomarkers and predictors of immunotherapy response. FGFR overexpression in gastric adenocarcinoma tissues correlates with poorer OS and PFS compared to normal gastric mucosa. Multivariate analyses identify FGFR4 as an independent prognostic indicator, with experimental models confirming that FGFR4 overexpression drives tumor cell proliferation, invasion, and migration. Immune profiling reveals divergent correlations: FGFR1 expression positively associates with infiltration of CD8+ T cells, CD4+ T cells, macrophages, and dendritic cells, while FGFR4 demonstrates a negative correlation with tumor-infiltrating lymphocytes. Functional studies in NOG mice show that FGFR1 overexpression enhances immunotherapy efficacy, whereas FGFR4 overexpression diminishes therapeutic response. Notably, combining FGFR4 inhibitors with anti-PD-1 therapy significantly enhances antitumor effects in FGFR4-overexpressing xenograft models. 126 The divergent correlations between FGFR isoforms and immune cell infiltration—with FGFR1 positively associated with CD8+ T-cell, macrophage, and dendritic cell infiltration, while FGFR4 demonstrates negative associations with tumor-infiltrating lymphocytes—carry significant clinical implications for patient selection and combination therapy strategies. These findings suggest that FGFR1-overexpressing tumors may already exist in a relatively immune-active microenvironment, potentially rendering them more responsive to immunotherapy approaches. Conversely, FGFR4-driven GC appear to create an immune-excluded or immune-desert phenotype, characterized by limited T-cell infiltration and resistance to single-agent PD-1 blockade. The preclinical observation that combining FGFR4 inhibitors with anti-PD-1 therapy synergistically enhances antitumor effects in xenograft models provides direct experimental support for this concept. 126 From a translational perspective, FGFR isoform profiling could serve as a predictive biomarker for immunotherapy response and guide rational combination strategies: FGFR1-high tumors might be prioritized for immunotherapy-containing regimens, while FGFR4-high tumors may require upfront combination of FGFR inhibition with immune checkpoint blockade to overcome immune exclusion. Furthermore, the differential immune modulation mediated by distinct FGFR family members highlights the importance of isoform-selective targeting strategies, as pan-FGFR inhibition might produce unpredictable net effects on the tumor immune microenvironment depending on the relative expression of individual FGFRs. These mechanistic insights position FGFR-targeted therapy not merely as a direct anti-proliferative strategy but as a potential immunomodulatory intervention that could be optimized through biomarker-guided patient selection and rational combination with immunotherapy. The predominant FGFR alterations in GC include FGFR1 mutations, FGFR2 amplifications, and FGFR3 rearrangements. 127 Clinical prevalence studies indicate FGFR2 positivity in 25.6% of GC patients (DS-Screen analysis, 2022), 128 while approximately 10% exhibit abnormal FGFR pathway activation. 129 These molecular characteristics and clinical observations collectively indicate substantial therapeutic potential for anti-FGFR2 strategies in gastric adenocarcinoma management.
Bemarituzumab represents the benchmark therapeutic targeting FGFR2 in GC. This humanized IgG1 mAb selectively binds the extracellular domain of the tumor-specific FGFR2b splice variant. 130 Initial clinical validation emerged from the 2020 FPA144 phase I trial, which established Bemarituzumab’s favorable safety profile and monotherapy activity in advanced gastric adenocarcinoma. 131 Subsequent 2021 FIGHT trial phase I data confirmed therapeutic benefit: combining Bemarituzumab with chemotherapy significantly prolonged PFS and OS in advanced FGFR2-positive G/GEJ cancer, though ocular adverse events were documented. 132 A 2022 randomized double-blind phase II study further demonstrated efficacy, with the Bemarituzumab cohort achieving 9.5 months of DFS versus 7.4 months in placebo controls. 133 The current investigation focuses on a phase III trial evaluating first-line Bemarituzumab-chemotherapy combinations in advanced GEJ adenocarcinoma patients with high FGFR2b expression. 131
The clinical utility of anti-FGFR therapies is frequently constrained by rapid resistance development, compromising efficacy and accelerating disease progression. 129 This limitation primarily stems from the reversible receptor binding mechanism characterizing most investigational FGFR inhibitors, rendering them vulnerable to acquired resistance. Futibatinib represents a distinct pharmacological class as an irreversible, highly selective inhibitor of FGFR1–4 that covalently modifies FGFR2. Its unique mechanism confers both durable target suppression and retained activity against kinase domain mutations conferring resistance to reversible inhibitors. 134 Initial clinical evaluation in a 2020 phase I trial established Futibatinib’s safety profile, pharmacodynamic activity, and preliminary efficacy in advanced solid tumors. 135 Subsequent 2022 phase I data confirmed antitumor activity with an ORR of 13.7%, 136 while a 2023 Japanese phase I study reported 11.5% ORR in patients with FGF/FGFR abnormalities. Notably, GC patients exhibiting high FGFR2 copy number (CN ⩾10) achieved 36.4% ORR versus no responses in CN < 10 subgroups. 137 These findings not only validate Futibatinib’s clinical feasibility but also highlight its exceptional efficacy in FGFR2-high amplified malignancies, suggesting biomarker-driven application merits further investigation.
Beyond established agents, novel FGFR-targeted therapeutics continue to emerge. The 2022-introduced RK-019 features a pyridopyrimidinone scaffold that potently inhibits FGFR1–4 phosphorylation and downstream signaling effectors (FRS2, PLCγ, AKT, Erk), inducing cell cycle arrest and apoptosis. This compound demonstrates potent anti-proliferative effects against FGFR2-amplified GC cells in vitro, while daily oral administration significantly suppresses xenograft growth without observable toxicity. 138 Concurrently, Pemigatinib is under evaluation in the phase II FiGhTeR trial (NCT05202236) for trastuzumab-resistant metastatic G/GEJ cancer, having demonstrated acceptable safety despite pending efficacy results. These continuous innovations in FGFR-targeted therapeutics are expanding the therapeutic arsenal for gastric adenocarcinoma through optimized target engagement and resistance management strategies. Resistance to FGFR-targeted therapies frequently limits their clinical utility, with mechanisms broadly classified into on-target and off-target alterations. On-target resistance primarily involves acquired mutations within the FGFR kinase domain that impair drug binding while preserving kinase activity. The most well-characterized are gatekeeper mutations, such as FGFR2 V564I (analogous to the “gatekeeper” residue in other kinases), which sterically hinder inhibitor access to the ATP-binding pocket without disrupting ATP binding itself. These mutations confer resistance to reversible ATP-competitive inhibitors while often remaining sensitive to irreversible inhibitors that form covalent bonds with cysteine residues. Other resistance mutations include molecular brake mutations (e.g., FGFR2 N549H/K) that destabilize the inactive kinase conformation required for inhibitor binding, and activation loop mutations that lock the kinase in a constitutively active state. Off-target resistance involves activation of alternative signaling pathways that bypass FGFR dependence, including upregulation of other RTKs (EGFR, MET, HER2) or downstream effectors (PI3K/AKT, MAPK). Epithelial-mesenchymal transition has also been implicated as a resistance mechanism, with mesenchymal phenotype associated with reduced FGFR dependence and sensitivity. Tumor heterogeneity complicates resistance management, as different metastatic lesions may harbor distinct resistance mechanisms. The development of next-generation FGFR inhibitors with activity against gatekeeper mutations (e.g., irreversible inhibitors like futibatinib, which maintain efficacy against V564I-mutant FGFR2) represents a key therapeutic advance. Ongoing research focuses on rational combination strategies to prevent or overcome resistance, including FGFR plus EGFR/MET inhibition and FGFR plus PI3K/AKT blockade, as well as liquid biopsy approaches to detect emerging resistance mutations before clinical progression.
Other targets
N6-methyladenosine
Epigenetics encompasses heritable alterations in gene expression independent of DNA sequence changes, including DNA methylation and chromatin conformational shifts. Among epigenetic regulators, m6A RNA modification represents the most prevalent and functionally significant internal methylation in mammalian mRNA. Dysregulated m6A dynamics are implicated in multiple oncogenic processes—including RNA processing, splicing, stabilization, and translational control—contributing to the pathogenesis and progression of various malignancies. 139 Growing evidence demonstrates that m6A-mediated RNA methylation actively participates in GC epigenetics, mechanistically influencing both carcinogenesis and tumor evolution.
m6A RNA modification is dynamically regulated by three protein classes: writers (methyltransferases), erasers (demethylases), and readers (recognition proteins). 139 The writer complex catalyzes m6A deposition, primarily through Methyltransferase-like 3 (METTL3) acting with METTL14, Wilms Tumor 1-associating protein, and the scaffolding protein KIAA1429. Conversely, erasers mediate m6A removal, with fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5) serving as primary enzymatic mediators. 140 Reader proteins selectively recognize m6A modifications without altering methylation status, facilitating downstream biological interpretation.
At present, m6A-related research in GC remains primarily in the prognostic biomarker and mechanistic exploration phases, with no approved therapies targeting m6A regulators currently available for clinical use. Among m6A regulatory proteins, the methyltransferase METTL3 has garnered the most research attention. METTL3 exerts influence over GC development by modulating critical processes like cellular growth, proliferation, metastatic spread, resistance to chemotherapy, and blood vessel formation. This oncogenic activity is mediated through several defined signaling cascades, including the METTL3-YAP1-Hippo, METTL3-PBX1-GCH1-BH4, METTL3-KRT18-MAPK/ERK, and METTL3-SPHK2-KLF2 axes. 141 Consequently, METTL3 functions as a driver oncogene in GC, where its frequent overexpression correlates with worsened patient outcomes and enhanced tumor aggressiveness-establishing its prognostic significance.
Beyond methyltransferases, demethylating enzymes also play crucial roles in GC pathogenesis. Elevated ALKBH5 levels in GC cells result in the demethylation of the long non-coding RNA NEAT1. 142 Functioning as a molecular scaffold, demethylated NEAT1 subsequently modulates the expression of EZH2 target genes implicated in promoting tumor cell invasion and metastatic capability. Thus, ALKBH5 overexpression facilitates the metastatic progression of GC, with lower ALKBH5 expression levels in GC tissue samples correlating with distant organ and lymph node metastases. 143
Currently, no ongoing clinical trials specifically targeting m6A regulators in GC are registered on ClinicalTrials.gov or major international trial registries. This reflects the early stage of the field and the substantial translational barriers that remain, including: (1) the essential physiological roles of m6A regulators in normal tissues, raising concerns about on-target toxicity; (2) the complexity of the m6A regulatory network, with multiple writers, erasers, and readers exhibiting context-dependent and sometimes opposing functions; (3) challenges in developing selective small molecule inhibitors that discriminate between closely related family members; and (4) the absence of validated predictive biomarkers to identify patients most likely to benefit.
Discussion
GC represents a widespread and deadly malignancy within the gastrointestinal tract, constituting a major worldwide health burden. Patients presenting with advanced-stage disease frequently encounter limited effective treatments, accentuating the critical demand for integrated therapeutic approaches. Targeted therapy has arisen as a viable strategy in recent years, providing new avenues to enhance outcomes for GC patients. This comprehensive analysis outlines the primary therapeutic targets and associated pharmacological agents in GC targeted therapy, seeking to deliver key insights for healthcare practitioners and steer future investigative priorities (Figure 4(a)).
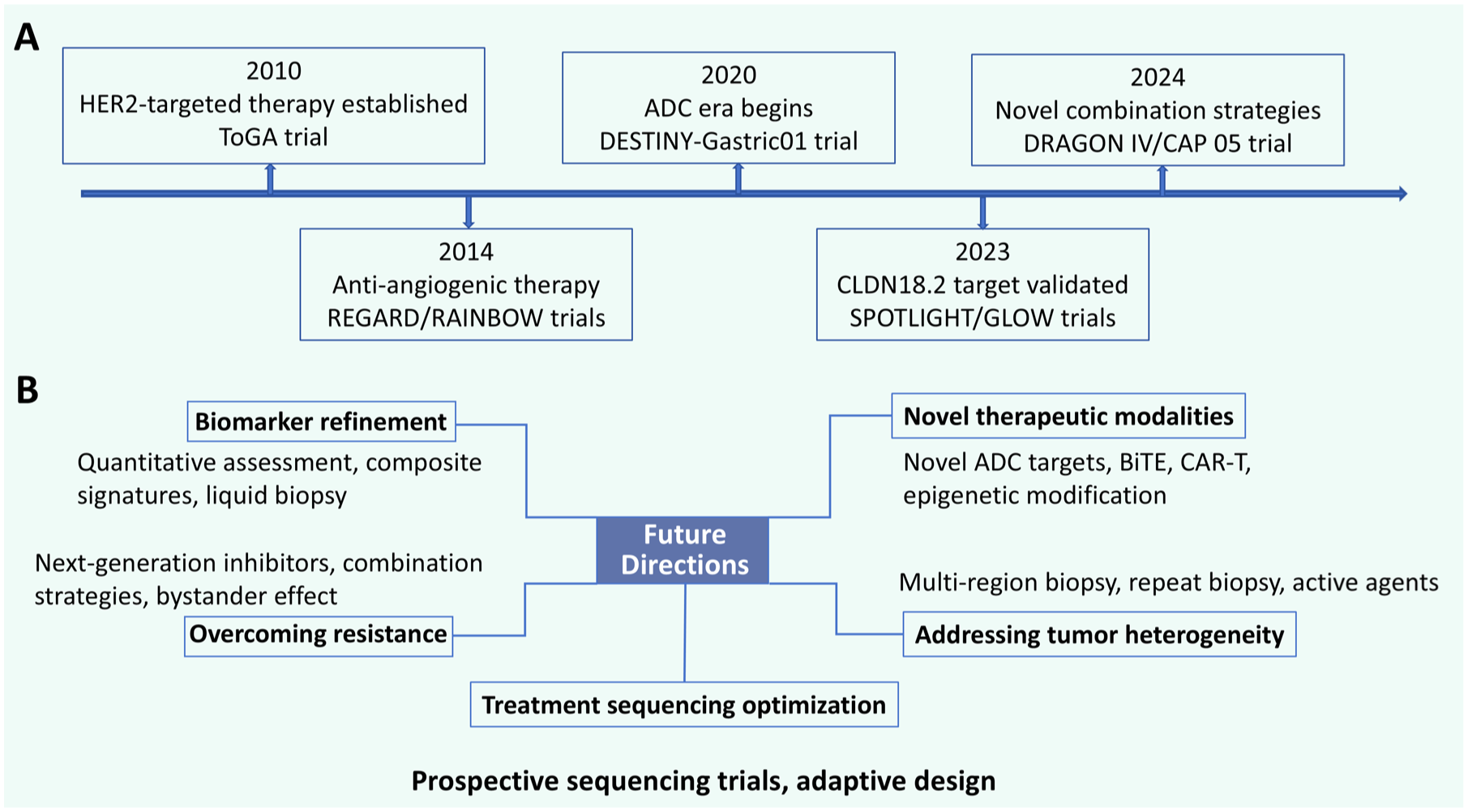
History and future perspectives of targeted therapy in gastric cancer. (a) Timeline of key milestones and pivotal clinical trials in the evolution of gastric cancer targeted treatments, from the establishment of HER2-targeted therapy to recent novel combination strategies. (b) Schematic representation of key future directions in gastric cancer research and clinical management, highlighting biomarker refinement, development of novel therapeutic modalities, strategies to address tumor heterogeneity and overcome resistance, as well as treatment sequencing optimization.
HER2 remains a validated therapeutic focus in GC management. Detectable in roughly 20% of GC cases, this target has driven substantial therapeutic innovation. The diversification of HER2-directed agents has meaningfully expanded clinical options. Monoclonal antibodies, exemplified by Trastuzumab, consistently deliver significant clinical benefit in both initial systemic treatment and perioperative settings. The landmark ToGA trial cemented Trastuzumab combined with chemotherapy as the standard first-line regimen for HER2-positive disease. Extensive subsequent trials have further corroborated its clinical utility across diverse therapeutic contexts. Margetuximab, with its optimized Fc region, shows potential as an alternative to Trastuzumab, especially in cases of Trastuzumab resistance. Pertuzumab targets a distinct extracellular domain of HER2 and initially showed controversial results in the JACOB trial. However, subsequent subgroup analyses and reanalysis have confirmed its therapeutic effectiveness and safety. Ongoing trials are expected to provide further evidence of its benefits. Bispecific formats targeting distinct HER2 domains, including ZW25 and KN026, display notable clinical activity against tumors, highlighting their potential to inspire next-generation drug design.
ADCs like T-DXd have broadened the application of HER2-targeted therapy by effectively treating patients with low HER2 expression. T-DXd has shown significant efficacy in several clinical trials, leading to its approval for clinical use in G/GEJ adenocarcinoma. Other ADCs, such as Disitamab vedotin (RC48) and ARX788, also show promise, although further research is needed to establish their effectiveness. On the other hand, TKIs targeting HER2, including Lapatinib, Tucatinib, and Pyrotinib, have encountered challenges regarding their efficacy. The effectiveness of Lapatinib in treating GC remains uncertain, while the clinical efficacy of Tucatinib in GC has not yet been thoroughly investigated. Pyrotinib shows potential in combination therapies and as an adjuvant to radiotherapy, but more research is required to optimize its use.
Targeting CLDN18.2 holds significant clinical potential for advancing GC therapy. Its high expression rate in GC patients holds the potential to benefit a larger proportion of patients. As the sole clinically approved CLDN18.2-directed agent, Zolbetuximab demonstrates substantial antitumor activity in monotherapy and combination regimens, establishing it as an emerging standard for defined molecular subgroups. CAR-CLDN18.2 T cells (CT041) have also demonstrated potential in treating G/GEJ adenocarcinoma, although challenges such as tumor heterogeneity and high cost need to be addressed.
Inhibition of the VEGF/VEGFR axis constitutes a cornerstone of anti-angiogenic strategies for GC. The anti-VEGFR-2 mAb RAM is guideline-endorsed for second-line management of advanced disease. Substantial survival improvements have been documented across trials exploring its integration with diverse chemotherapy backbones and immunotherapies. Similarly, the orally bioavailable VEGFR-2 TKI Apatinib demonstrates clinically meaningful activity both as monotherapy and within combinatorial regimens. Other novel anti-angiogenic agents such as Fruquintinib, BC001, Vandetanib, and Sorafenib are being explored, each with its unique mechanism of action and potential in GC treatment.
FGFRs, associated with immune cell infiltration in GC, have become a critical target. Bemarituzumab, a humanized IgG1 mAb targeting FGFR2b, has shown activity in treating advanced FGFR2-positive G/GEJ cancer, although side effects such as eye problems need to be managed. The irreversible pan-FGFR inhibitor Futibatinib exhibits particular promise in GC patients exhibiting elevated FGFR2 copy number. Novel agents such as RK-019 and Pemigatinib are undergoing active investigation, reflecting sustained advancement in FGFR-directed therapeutics for GC. Concurrently, dysregulation of m6A RNA methylation functions as a critical epigenetic modulator in gastric carcinogenesis. Key regulatory factors, including METTL3 and ALKBH5, drive oncogenic progression through diverse signaling cascades, positioning them as promising candidates for next-generation therapeutic development. However, at present, there are no specific drugs on the market targeting these pathways, and further research is needed to translate these findings into clinical applications.
While targeted therapies have markedly advanced GC management, formidable hurdles continue to impede optimal patient outcomes. The emergence of drug resistance, a recurrent issue exemplified by FGFR inhibitors, constitutes a primary limitation. Tumor heterogeneity further complicates therapeutic interventions, particularly hindering the application of adoptive cellular therapies such as CAR-T in solid tumors like GC. Compounding these biological challenges, the considerable expense associated with advanced modalities, including CAR-T technology, restricts their broad clinical accessibility. Addressing these constraints necessitates focused future endeavors: developing next-generation agents with intrinsic resistance evasion capabilities, enhancing therapeutic strategies to overcome intratumoral heterogeneity, and implementing measures to improve treatment affordability. Concurrently, advancing biomarker-driven stratification is paramount to accurately identify patient subsets most responsive to specific targeted agents. This precision approach will refine therapeutic decision-making and ultimately elevate the OS trajectory for GC patients.
Collectively, the preceding discussion underscores a paradigm shift in our understanding of targeted therapies: beyond their direct tumor-suppressive effects, these agents actively reshape the tumor immune microenvironment, creating opportunities for rational integration with immunotherapy. HER2-targeted antibodies enhance antigen presentation and T-cell priming; VEGF/VEGFR inhibition relieves immunosuppressive barriers and facilitates lymphocyte infiltration; FGFR isoform-selective targeting modulates immune cell recruitment in opposing directions. This immunomodulatory dimension of targeted therapy has profound clinical implications. First, it argues against viewing targeted therapy and immunotherapy as independent treatment modalities—rather, their biological effects are complementary and potentially synergistic when properly sequenced or combined. Second, it highlights the importance of dynamic biomarker assessment that captures not only target expression but also immune contexture, as the optimal treatment strategy may depend on both tumor cell genetics and the state of the tumor immune microenvironment. Third, it suggests that the full therapeutic potential of many targeted agents may only be realized when combined with immunotherapy in appropriately selected patient populations. Future research should prioritize mechanistic studies elucidating how individual targeted therapies modulate specific immune cell subsets, clinical trials designed to test rational combination and sequencing strategies, and development of composite biomarkers that integrate genomic and immunologic parameters to guide precision oncology decisions in GC.
The distinction between prognostic and predictive biomarkers has important implications for the interpretation of clinical trial data and the translation of research findings into clinical practice. Across the targeted therapy landscape in GC, we observe varying relationships between biomarker expression and clinical outcomes. For HER2, the biomarker serves both prognostic and predictive roles: HER2 amplification confers aggressive tumor biology and worse prognosis without targeted intervention, yet identifies patients who derive substantial benefit from HER2-directed therapies. For CLDN18.2, the prognostic association appears favorable—expression correlates with improved outcomes independent of treatment—while the predictive value for zolbetuximab response remains robust and clinically meaningful. For FGFR2, current evidence primarily supports its predictive role for agents such as bemarituzumab and futibatinib, while its independent prognostic significance requires further investigation. For VEGF/VEGFR, expression patterns are primarily predictive for anti-angiogenic agent efficacy, with limited independent prognostic value. These distinctions are not merely academic: they influence how clinicians interpret biomarker test results, how trials are designed and stratified, and how regulatory decisions are made regarding drug approvals and label indications. Future biomarker research should explicitly characterize both prognostic and predictive components, enabling more nuanced integration of molecular information into clinical decision-making algorithms.
The recognition of intratumoral heterogeneity has profound implications for biomarker-driven patient selection in clinical practice. Current standard practice relies on single-site biopsy specimens obtained at diagnosis to determine HER2, CLDN18.2, or FGFR2 status. However, given the spatial heterogeneity documented in GC, a single biopsy may not adequately represent the molecular landscape of the entire tumor burden. This sampling limitation can lead to false-negative classification, denying potentially effective targeted therapy to patients whose tumors harbor actionable alterations in unsampled regions. Moreover, temporal heterogeneity—with biomarker status potentially evolving during disease progression or under selective treatment pressure—raises questions about the validity of archival tissue for guiding later-line therapeutic decisions. Repeat biopsies at progression, though not routinely performed, may provide valuable information about clonal evolution and acquired resistance mechanisms. The advent of ADCs with bystander killing capacity partially mitigates these concerns by enabling efficacy even in heterogeneous expression settings, potentially broadening the population that may benefit from targeted approaches. Nevertheless, optimizing biomarker assessment strategies—through multi-region sampling, liquid biopsy approaches detecting circulating tumor DNA, or development of sensitive imaging biomarkers—remains a priority to fully realize the potential of precision oncology in GC.
Furthermore, addressing tumor heterogeneity requires a deep understanding of the intersection between histopathological subtypes and molecular phenotypes. GC exhibits profound morphological diversity, frequently presenting with varying percentages of signet ring cell (SRC) components in poorly cohesive or mixed-type carcinomas. Recent literature underscores that the specific proportion of SRCs significantly impacts clinical outcomes; for instance, the percentage of SRCs has been shown to be inversely related to aggressive behavior and poor prognosis in mixed-type GC, 144 and it serves as a potential novel predictor of survival in poorly cohesive GC patients. 145 To further stratify these complex patient risk profiles and guide clinical decision-making, novel diagnostic markers, such as serum CD26 levels, are also being actively explored. 146 This profound histopathological heterogeneity has direct and vital implications for targeted therapeutic responsiveness, as molecular alterations are frequently bound to specific histologic subtypes. For example, HER2 positivity demonstrates a strong preponderance for intestinal-type GC and typically correlates inversely with SRC-rich or poorly cohesive histologies. Consequently, patients whose tumors harbor significant SRC components often derive limited therapeutic benefit from standard anti-HER2 monoclonal antibodies. Conversely, CLDN18.2 expression correlates significantly with the Lauren diffuse type, which inherently encompasses many SRC and poorly cohesive morphologies. This distinct distribution positions CLDN18.2 as an exceptionally relevant therapeutic target for diffuse and SRC histologies, bridging a critical therapeutic gap where HER2-targeted interventions traditionally fall short. Moreover, in these highly heterogeneous histomolecular subgroups, ADC strategies become increasingly pertinent. Because mixed-type and SRC-containing GC frequently display uneven molecular landscapes, therapies requiring uniform target expression often fail. The unique “bystander effect” of ADCs—allowing the cytotoxic payload to diffuse and eradicate adjacent antigen-negative or low-expressing tumor cells—offers a distinct mechanistic advantage. This enables ADCs to effectively overcome the spatial heterogeneity and variable target expression inherent to specific histological subtypes, presenting a highly relevant strategy for mixed-type and SRC-rich tumors.
Future perspectives
The therapeutic landscape for advanced GC has evolved substantially over the past decade, yet significant challenges remain, and numerous unanswered questions warrant continued investigation. Based on the evidence synthesized in this review, we propose several key directions for future research and clinical development (Figure 4(b)).
First, biomarker-driven precision oncology must be refined beyond single-gene testing. The success of HER2-directed therapies, CLDN18.2 targeting with zolbetuximab, and FGFR2b inhibition with bemarituzumab demonstrates that molecular stratification can identify patient populations with enhanced therapeutic benefit. However, the current paradigm of binary biomarker positivity (e.g., HER2-positive vs HER2-negative) may oversimplify biologically complex tumors. Future approaches should incorporate quantitative biomarker assessment (e.g., HER2 copy number, CLDN18.2 expression levels), spatial and temporal heterogeneity, and composite biomarker signatures that integrate genomic, transcriptomic, and immunologic parameters. The development of liquid biopsy technologies enabling real-time monitoring of biomarker evolution and early detection of resistance mutations will be critical for dynamic treatment adaptation.
Second, overcoming therapeutic resistance requires mechanistic insights and combinatorial strategies. As detailed in this review, resistance mechanisms span on-target alterations (e.g., HER2 kinase domain mutations, FGFR gatekeeper mutations), bypass pathway activation (e.g., MET amplification, PI3K/AKT upregulation), microenvironmental adaptations, and immune evasion. Future drug development should prioritize next-generation agents designed to circumvent common resistance mechanisms—such as irreversible FGFR inhibitors active against gatekeeper mutations (e.g., futibatinib), antibody-drug conjugates with bystander effects that overcome heterogeneous target expression (e.g., T-DXd), and BsAbs engaging multiple epitopes or immune effectors. Rational combination strategies targeting parallel pathways (e.g., HER2 plus MET inhibition, FGFR plus EGFR blockade) and integrating targeted agents with immunotherapy (e.g., HER2 blockade plus PD-1 inhibition, VEGF inhibition plus checkpoint blockade) hold promise for extending response durability and delaying resistance emergence.
Third, optimal sequencing and combination strategies must be defined through dedicated trials. The observation that prior immunotherapy exposure enhances subsequent RAM efficacy illustrates the importance of treatment sequencing—a dimension often overlooked in traditional trial designs. Future studies should prospectively evaluate not only whether to combine agents but also the optimal order, duration, and timing of interventions. Adaptive trial designs incorporating biomarker-driven treatment switches at progression may identify personalized sequencing algorithms that maximize cumulative benefit. Furthermore, the integration of targeted therapy into perioperative and adjuvant settings—where curative intent creates higher stakes for both efficacy and toxicity—requires rigorous evaluation in randomized trials with long-term follow-up.
Fourth, novel therapeutic modalities and targets warrant accelerated investigation. Beyond the established targets reviewed here, emerging classes of agents are entering clinical evaluation. Antibody-drug conjugates targeting novel antigens (e.g., TROP2, B7-H3, HER3) are in early-phase trials; bispecific T-cell engagers and CAR T-cell therapies are being refined for solid tumor applications; and epigenetic modifiers targeting m6A regulators (e.g., METTL3 inhibitors) are advancing toward clinical translation. Although the m6A field remains primarily mechanistic and prognostic at present, the identification of targetable downstream effectors (e.g., ferroptosis regulators, immune-modulatory lncRNAs) suggests that epigenetic vulnerabilities may eventually become actionable. Investment in preclinical models that recapitulate tumor heterogeneity and the tumor microenvironment will be essential for prioritizing the most promising candidates for clinical development.
Fifth, addressing tumor heterogeneity—both intratumoral and intertumoral—requires innovative approaches. The spatial and temporal heterogeneity documented for HER2, CLDN18.2, and FGFR2 amplifications challenges the reliance on single-site archival biopsies for treatment decisions. Strategies to mitigate heterogeneity include: (1) multi-region sampling at diagnosis to capture clonal architecture; (2) repeat biopsies at progression to assess clonal evolution; (3) liquid biopsy-based monitoring for early detection of resistant subclones; and (4) development of therapeutic agents with activity against heterogeneous target expression, such as ADCs with bystander killing effects. Future trials should incorporate translational substudies that systematically characterize heterogeneity and correlate it with treatment outcomes.
Finally, global equity in access to precision oncology must be prioritized. The GC burden is disproportionately high in Asia, Latin America, and Eastern Europe, yet access to biomarker testing and targeted therapies remains uneven. Efforts to develop cost-effective companion diagnostics, generic or biosimilar versions of approved agents, and simplified treatment regimens suitable for resource-limited settings are essential to ensure that advances in precision oncology benefit patients worldwide. Collaborative international trials and public-private partnerships will be critical to achieving this goal.
In conclusion, the future of GC treatment lies in the convergence of deeper biological understanding, innovative therapeutic platforms, and personalized strategies that account for tumor heterogeneity and dynamic evolution. By addressing the challenges outlined above-refining biomarkers, overcoming resistance, optimizing sequencing, exploring novel modalities, tackling heterogeneity, and ensuring equitable access—the field can build upon the foundational advances of the past decade to meaningfully improve outcomes for patients with this devastating disease.