
Abstract
Inflammatory bowel disease (IBD), including ulcerative colitis and Crohn's disease, is characterized by chronic nonspecific intestinal inflammation. Despite considerable efforts, IBD remains a heavy burden on society and human health, with increasing morbidity. Posttranslational modification, especially histone acetylation, is a key process in controlling DNA transcriptional activity. Histone deacetylases (HDACs) play a vital role in the mechanism of IBD pathogenesis through histone and nonhistone protein deacetylation. Herein, we present a summary of different categories of HDACs as well as HDAC inhibitors (HDACis) and analyze the role of HDAC inhibition in alleviating IBD along with its mechanism, as well as clinical potential of HDACis in IBD treatment.
Keywords
Introduction
Inflammatory bowel disease (IBD) is a general term for a group of diseases characterized by chronic nonspecific intestinal inflammation, mainly ulcerative colitis (UC) and Crohn's disease (CD). Lesions in UC are usually confined to the colonic mucosa, with a common expansion pattern from the rectum to the proximal colon, and persistent superficial inflammation is frequently accompanied by ulcers. 1 CD can affect the whole digestive tract from the oral cavity to the anus, with lesions distributed in a segmental and jumping pattern. The terminal ileum and ileocecal junction are the most common sites of CD occurrence. In addition to common manifestations, such as abdominal pain, diarrhea, and hematochezia in UC, CD can also lead to perianal lesions, intestinal stenosis, fistulas, etc. 2
Epidemiology studies have shown that the incidence and prevalence of IBD have increased rapidly worldwide in recent years, especially in newly industrialized areas such as Asia. 3 In China, from 1990 to 2019, the female age-standardized prevalence rate increased from 20.7 cases per 100,000 persons (95% uncertainty interval (UI) 17.2–24.5) to 44.2 cases per 100,000 persons (95% UI 37.7–51.4), with an annual rate of change of 1.14% (95% CI 1.05–1.26); the male age-standardized prevalence rate increased from 25.1 cases (95% UI 20.9–30.2) to 50.0 cases (95% UI 42.5–58.5) per 100,000 population, with an annual rate of change of 0.99% (95% CI 0.90–1.09). 4 As a chronic and incurable disease, IBD impedes both the physical and psychological health of patients, and it is speculated that the direct medical costs attributable to IBD patients are three times greater than those associated with patients without IBD and are even increasing these years, placing a heavy burden on both families and the society as a whole. 5
To date, the pathogenesis and etiology of IBD are still not clear and are believed to be related to multiple factors, including genetics, immunity, the environment and gut microorganisms, etc. 6 Despite many choices of medications, such as aminosalicylates, corticosteroids, and immunosuppressants, 7 a portion of patients still suffer from unsatisfactory therapeutic effects, disease relapse and withdrawal of medication due to side effects (including infections, anemia, headaches, lymphopenia, and cardiovascular events). 8 Surgery is ultimately inevitable among patients who are not responsive to medication, and for CD, the recurrence rate even after surgery approaches 100%, often at the anastomosis. 9 In summary, IBD places a heavy burden on healthcare services, and new effective medications with minimal side effects are urgently needed.
In recent years, experts in IBD drug development have focused on a new promising field, epigenetics, a new regulatory mechanism of diseases. 10 Epigenetics refers to the control of gene expression by altering chromatin, not the DNA sequence. 11 In eukaryotic cells, nucleosomes are the basic subunits of chromatin, and their core particles are formed by genomic DNA superhelices wrapped around histone octamers.12–14 Histones, as the main components of chromatin, can be divided into five types, H2A, H2B, H3, H4, and H1, which promote gene transcription when DNA and histones are loosely attached. 15 The N- and C-termini of core histones mediate the high-level folding of chromatin, and the different amino acids in their tails (such as lysine, arginine, serine, and threonine) can be targeted for posttranslational modification (PTM), which regulates DNA binding to transcription factors and RNA polymerase II and then causes transcription to cease because of compact chromatin. 16
PTMs of histones include methylation, acetylation, phosphorylation, ubiquitination, malonylation, propionylation, butyrylation, crotonylation, and lactylation.17–20 PTMs can participate in IBD pathogenesis by modulating immune cells, cytokine secretion, the intestinal flora, etc.21–23 Among all PTMs, histone acetylation is the most common.24–26 Histone acetylation is regulated by histone deacetylase (HDAC) and histone acetylase (HAT): HATs catalyze the translocation of acetyl groups to lysine residues, which loosens chromatin for transcription, whereas HDACs deacetylate histones, resulting in stronger attachment to DNA, compressed chromatin, and reduced transcriptional activity. 27 Evidence has indicated that histone acetylation is promoted, and HDAC inhibitor (HDACi) administration relieves disease severity in IBD. 28 Therefore, in this review, we will systematically review the effects and mechanisms of HDACs in IBD and discuss the potential clinical application and future prospects of HDACis in IBD treatment. The summarized frame of current review is shown in Figure 1.
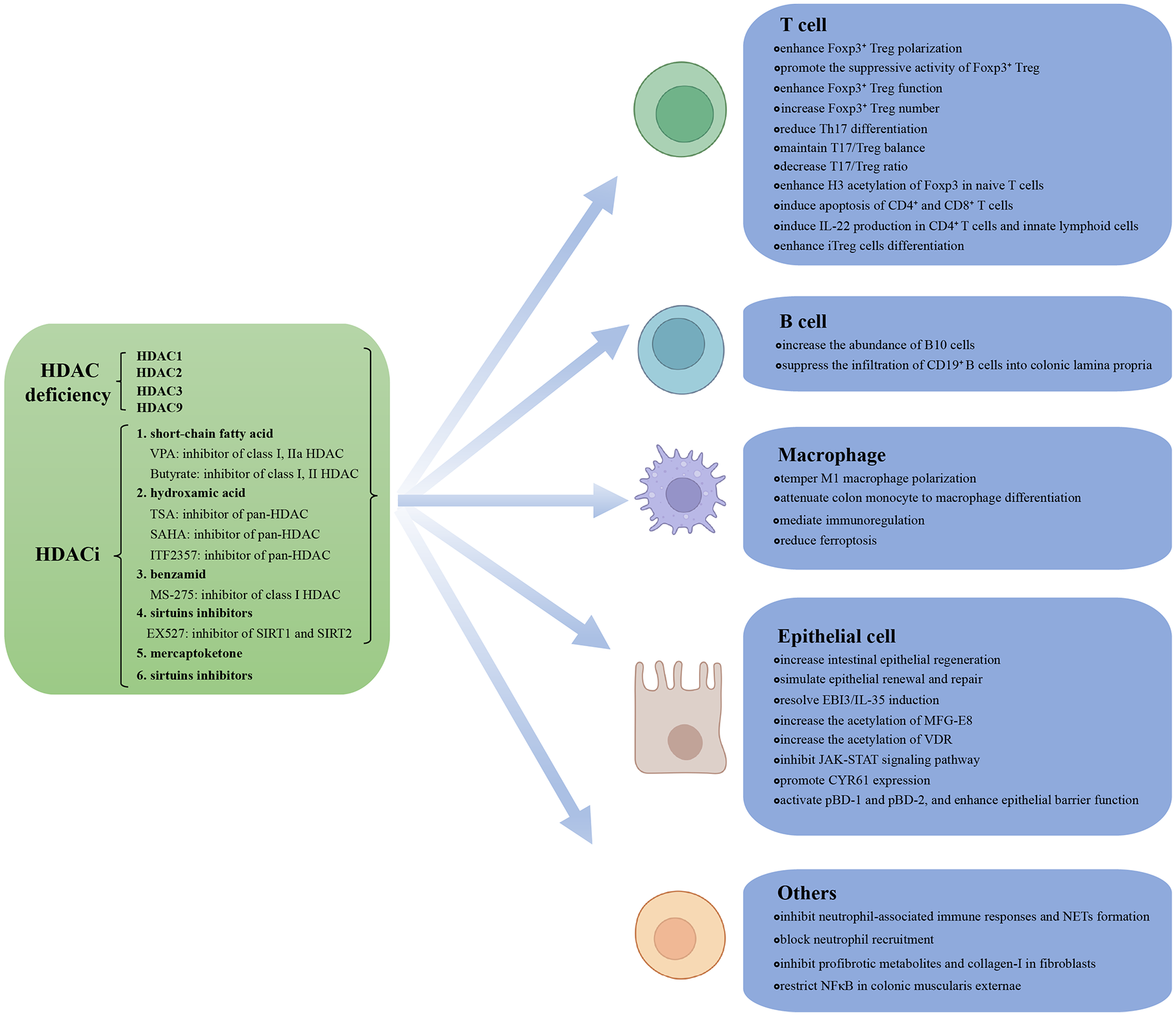
Summary of the mechanisms by which HDACis and HDAC deficiency inhibit IBD. HDAC deficiency and HDACis inhibit IBD through regulating T cell, B cell, macrophage, epithelial cell, and other cells function, respectively.
Histone deacetylases
On the basis of their structure, there are 18 different isoforms of HDACs that belong to four different classes29–31: class I Rpd3-like proteins (HDAC1, HDAC2, HDAC3, and HDAC8); class II Hda1-like proteins (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10); class III Sir2-like proteins (SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, and SIRT7); and class IV proteins (HDAC11). Class I, II, and IV HDACs share similar structures and require zinc for catalysis, whereas class III HDACs require NAD as a cofactor for enzymatic activity. 32
Class I HDACs are highly homologous to the yeast HDAC Rpd3 and consist of a highly conserved deacetylation structural domain; they are expressed mainly in the nucleus and have strong deacetylation activity toward histones. 33 Among class I HDACs, HDAC1 is the most extensively studied and plays an important regulatory roles in angiogenesis, redox homeostasis and inflammatory signaling, 34 especially in regulating the Fas promoter region of activated T cells and mediating immune homeostasis. T cell dysfunction is linked to various autoimmune diseases, including IBD, rheumatoid arthritis, multiple sclerosis, and systemic lupus erythematosus.35,36 Dual HDAC1/HDAC2 loss has been reported to reduce the colonic inflammatory response. 37
HDAC inhibitors
HDACis have strong anti-inflammatory effects, and the application of HDACis in IBD has attracted increased attention in recent years. On the basis of their chemical structure, HDACis are categorized into seven types: short-chain fatty acids (SCFAs) (valproic acid/VPA, sodium butyrate, etc.), hydroxamic acids (trichostain-A/TSA, vorinostat/SAHA, givinostat/ITF2357, etc.), benzamides (entinostat/MS-275, chidamide, etc.), cyclic peptides (depsipeptide and apicidin), mercaptoketones (KD5170), sirtuin inhibitors (nicotinamide, sirtinol, EX527, etc.), and others (citarinostat, etc.).38,39 To date, the U.S. Food and Drug Administration (FDA) has approved five HDACis for the treatment of different diseases: vorinostat for cutaneous T cell lymphoma (CTCL); romidepsin for peripheral T cell lymphoma (PTCL) and CTCL; belinostat for PTCL; panobinostat for multiple myelomas (although withdrawn by the FDA in 2022); and givinostat for Duchenne muscular dystrophy.39–44 A variety of HDACis, such as VPA or SAHA,45,46 givinostat, 47 the statin hydroxamate, 48 and MS-275, 49 have also shown potential therapeutic effects in IBD.36,50,51 The role and mechanisms of action of HDACis in IBD will be discussed in depth.
Regulatory mechanism of HDACs in IBD
HDACs regulate T cell function
Multiple types of primary immune cells, including T cells,52,53 B cells, 54 macrophages, 55 and neutrophils,56,57 are generally acknowledged to be involved in IBD pathogenesis and serve as therapeutic targets. Among them, T cells have been shown to be the most important component mediating the regulation of histone acetylation in IBD (Table 1).
Summary of the mechanisms by which HDACs regulate different cells in IBD.
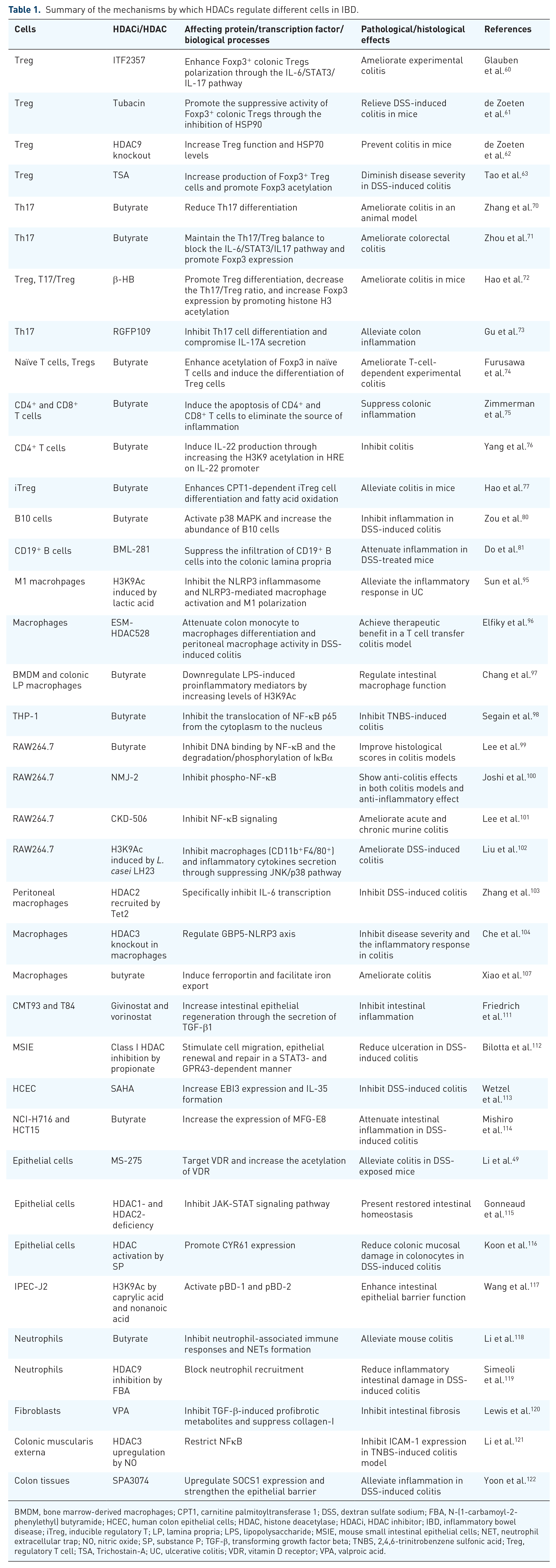
BMDM, bone marrow-derived macrophages; CPT1, carnitine palmitoyltransferase 1; DSS, dextran sulfate sodium; FBA, N-(1-carbamoyl-2-phenylethyl) butyramide; HCEC, human colon epithelial cells; HDAC, histone deacetylase; HDACi, HDAC inhibitor; IBD, inflammatory bowel disease; iTreg, inducible regulatory T; LP, lamina propria; LPS, lipopolysaccharide; MSIE, mouse small intestinal epithelial cells; NET, neutrophil extracellular trap; NO, nitric oxide; SP, substance P; TGF-β, transforming growth factor beta; TNBS, 2,4,6-trinitrobenzene sulfonic acid; Treg, regulatory T cell; TSA, Trichostain-A; UC, ulcerative colitis; VDR, vitamin D receptor; VPA, valproic acid.
Colonic regulatory T cells (Tregs) are characterized by the expression of transcription factor Foxp3 and are critical for maintaining intestinal homeostasis as well as limiting intestinal inflammation.58,59 The HDACi ITF2357 enhances Foxp3+ colonic Tregs polarization in the lamina propria (LP) through the IL-6/STAT3/IL-17 pathway, ameliorating experimental colitis. 60 The HDAC6-specific inhibitor tubacin has been shown to promote the suppressive activity of Foxp3+ colonic Tregs through the inhibition of heat shock protein 90 (HSP90), relieving dextran sulfate sodium (DSS)-induced colitis in mice. 61 HDAC9 knockout has been shown to increase Treg function and HSP70 levels and prevent colitis in mice. 62 In addition, HDACi TSA intervention has been shown to increase the number of Foxp3+ Treg cells and promote Foxp3 acetylation, diminishing disease severity in DSS-induced colitis. 63
In addition to Tregs, Th17 cells also participate in the pathogenesis of IBD and act as effector cells of HDACs during colitis.64–67 The intestinal mucosal immune microenvironment is regulated via cytokines, such as IL-26, IL-17, and IL-21.68,69 Butyrate has been shown to inhibit HDAC3 to reduce Th17 differentiation and ameliorate colitis in an animal model. 70 Butyrate has also been shown to maintain the Th17/Treg balance by inhibiting HDAC1 to block the IL-6/STAT3/IL17 pathway and promote Foxp3 expression, ultimately ameliorating colorectal colitis. 71 β-hydroxybuty rate (β-HB) has been shown to promote Treg differentiation, decrease the colonic Th17/Treg ratio, and increase Foxp3 expression by increasing histone H3 acetylation, ameliorating colitis in mice. 72 RGFP109, a selective inhibitor of HDAC1 and 3, has been shown to alleviate colon inflammation through the inhibition of Th17 cell differentiation from human peripheral blood mononuclear cells (PBMCs) and compromising IL-17A secretion. 73
Other mechanisms by which HDACs regulate T cells, such as naïve T cells, CD4+ and CD8+ T cells, and inducible regulatory T (iTreg) cells, also exist, as discussed below. Butyrate treatment enhances histone H3 acetylation in the conserved noncoding sequence regions and the promoter of the Foxp3 locus in naïve T cells, and induces the differentiation of Treg cells, ameliorating T-cell-dependent experimental colitis. 74 Butyrate suppresses colonic inflammation through a double-hit mechanism: inducing the apoptosis of CD4+ and CD8+ T cells to eliminate the source of inflammation and suppressing IFN-γ-induced STAT1 activation and inflammation. 75 Furthermore, butyrate can also induce IL-22 production in CD4+ T cells and innate lymphoid cells through increasing the H3K9 acetylation of the hypoxia response element on the IL-22 promoter, thus inhibiting colitis. 76 Butyrate also enhances carnitine palmitoyltransferase 1-dependent fatty acid oxidation and iTreg cells differentiation, alleviating DSS-induced colitis in mice. 77
HDACs regulate B cell function
Recent reports have focused on the important role of B lymphocytes in maintaining gastrointestinal homeostasis78,79 through HDAC inhibition (Table 1). B10 cells are regulatory B cells capable of producing IL-10 to maintain immune homeostasis, and a butyrate-induced increase in B10 cells has been observed in mice with DSS-induced colitis. The adoptive transfer of B cells pretreated with butyrate inhibits inflammation in DSS-induced colitis; mechanistically, transcriptomic analysis revealed that the activation of p38 MAPK by butyrate in an HDAC inhibitory activity-dependent manner is critical for increasing the abundance of B10 cells. 80 Moreover, the administration of a potent HDAC6 inhibitor, BML-281, has been shown to attenuate inflammation by suppressing the infiltration of CD19+ B cells into the colonic LP in DSS-treated mice, suggesting that targeting HDAC6 is an anti-inflammatory strategy against IBD progression. 81
HDACs regulate macrophage function
Macrophages are involved in both the initiation and progression of IBD through multiple mechanisms, including polarization,82–85 immunoregulation, 86 ferroptosis, 87 and pyroptosis,88,89 etc. Evidence has shown that histone deacetylation mediates the mechanism of which macrophage regulates in colitis (Table 1).
When stimulated by different internal environments or cytokines, M0 macrophages polarize into several phenotypes (including proinflammatory M1 macrophages and anti-inflammatory M2 macrophages), and the balance between M1 and M2 macrophages plays a critical role in intestinal homeostasis during IBD pathogenesis.90–94 HDAC inhibition has been reported to alleviate IBD through regulating macrophage polarization: histone 3 lysine 9 (H3K9) acetylation (H3K9Ac) induced by lactic acid has been shown to inhibit the NLRP3 inflammasome and NLRP3-mediated macrophage activation and M1 polarization, alleviating the inflammatory response in UC 95 ; and esterase-sensitive motif (ESM)-tagged HDACi ESM-HDAC528 has been shown to attenuate colon monocyte to macrophage differentiation and peritoneal macrophage activity in DSS-induced colitis, and achieve therapeutic benefit in a T cell transfer colitis model. 96
Macrophages mediate immunoregulation not only through the production of proinflammatory and anti-inflammatory cytokines, toxic mediators, chemokines, and macrophage extracellular traps but also via the modulation of epithelial cell proliferation and fibrosis in the intestine as well as its accessory tissues and the mediation of HDACs during these processes, especially the NF-κB signaling pathway. 86 Butyrate downregulates lipopolysaccharide (LPS)-induced proinflammatory mediators at the transcription level in bone marrow-derived macrophages and colonic LP macrophages, including nitric oxide, IL-12 and IL-6, by increasing levels of H3K9Ac. 97 In addition, butyrate has been shown to inhibit the LPS-induced translocation of NF-κB p65 from the cytoplasm to the nucleus in THP-1 monocytes and in PBMCs, and NF-κB p65 has also been shown to be inhibited by butyrate in 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis. 98 DNA binding by NF-κB has been shown to be inhibited, as has the degradation/phosphorylation of IκBα, in LPS-stimulated murine RAW264.7 cells (a macrophage cell line) by butyrate; histological scores in colitis models have also been shown to be improved after the administration of butyrate. 99 A cinnamyl sulfonamide hydroxamate derivative possessing nonselective HDAC inhibition ability, NMJ-2, has shown anti-colitis effects in both acetic acid and 2,4-dinitrochlorobenzene (DNCB)-induced colitis models and anti-inflammatory effects through the inhibition of phospho-NF-κB in LPS-induced RAW264.7 cells. 100 NF-κB signaling in macrophages and intestinal epithelial cells has been shown to be inhibited by CKD-506, a novel HDAC6 inhibitor, and to ameliorate acute and chronic murine colitis. 101
Other mechanisms are also involved in HDAC inhibition and macrophage-mediated immunoregulation in IBD. First, the restoration of H3K9Ac by Lactobacillus casei (L. casei) LH23 has been shown to ameliorate DSS-induced colitis by inhibiting macrophages (CD11b+F4/80+) as well as the secretion of inflammatory cytokines through suppressing JNK/p38 signal pathways and inducing an increase in CD3+CD4+CD25+ Tregs. 102 Second, ten eleven translocation (Tet) 2 has been shown to recruit HDAC2 and specifically inhibit IL-6 transcription through histone deacetylation in innate myeloid cells (including peritoneal macrophages and dendritic cells), and Tet2-deficient mice has been shown to be more susceptible to colitis. 103 Third, HDAC3 knockout in macrophages has been shown to inhibit disease severity and the inflammatory response in colitis by regulating the guanylate-binding protein 5 (GBP5)-NLRP3 axis. 104
Ferroptosis is a distinct form of iron-dependent nonapoptotic regulated cell death associated with IBD pathogenesis and interestingly, ferroptosis also participates in HDAC-mediated regulation of macrophages in IBD in a cell-specific way.87,105,106 Butyrate has also been shown to induce ferroportin expression in macrophages by decreasing the enrichment of HDACs around the Slc40a1 promoter, facilitating iron export and thus ameliorating colitis. 107
HDACs regulate epithelial cell function
Various types of epithelial cells, including Paneth, M, columnar, tuft and goblet cells, collaborate to maintain intestinal barrier integrity and play indispensable roles in regulating interactions between the immune system, the intestinal contents and other components.108,109 Recent studies have indicated that epithelial cell dysfunction is strongly associated with IBD pathogenesis 110 and mediates the regulation of histone deacetylation in IBD (Table 1).
Multiple HDACis have been shown to play a protective role in the intestinal epithelium of individuals with colitis: givinostat and vorinostat have been shown to increase intestinal epithelial regeneration through the secretion of transforming growth factor beta 1 (TGF-β1) in murine (CMT93) and human (T84) colonic epithelial cell lines, and givinostat has also been shown to improve epithelial wound healing and barrier recovery in DSS-stressed mice 111 ; propionate, a type of SCFA, has been shown to stimulate cell migration, epithelial renewal and repair upon class I HDAC inhibition, in a STAT3- and GPR43-dependent manner in mouse small intestinal epithelial cells, and propionate has also been shown to reduce ulceration in DSS-induced colitis 112 ; SAHA treatment has been shown to increase Epstein-Barr virus-induced gene 3 (EBI3) expression and anti-inflammatory cytokine IL-35 formation under inflammatory conditions in human colon epithelial cells, and inhibit DSS-induced colitis in wild-type but not in EBI3-deficient mice 113 ; and butyrate acid has been reported to increase the acetylation of H3K9 around the promoter of milk fat globule-epidermal growth factor 8 (MFG-E8) and the expression of MFG-E8 in colon epithelial cells (NCI-H716 and HCT15) and attenuate intestinal inflammation in DSS-induced colitis. 114
Our previous study demonstrated that the level of histone H3 acetylation was decreased in epithelial cells in UC patients and that the HDACi MS-275 targeted the vitamin D receptor (VDR) and increase the acetylation of VDR in epithelial cells to reduce inflammation and apoptosis, and restore intestinal barrier function, thereby alleviating colitis in DSS-exposed mice. 49
Genetic manipulation of or chemical intervention to disrupt HDACs or histone acetylation has also been shown to produce similar results: intestinal epithelial cell-specific HDAC1- and HDAC2-deficient mice have been shown to present restored intestinal homeostasis via the inhibition of the JAK-STAT signaling pathway 115 ; substance P (SP) has been shown to promote the cysteine-rich angiogenic inducer 61 (CYR61) expression through increased HDAC activity, and colonic mucosal damage has been shown to be reduced in colonocytes in DSS-induced mice 116 ; and the facilitation of the acetylation of H3K9 at the promoters of porcine β-defensins 1 (pBD-1) and pBD-2 by medium chain fatty acids (caprylic acid and nonanoic acid) has been shown to significantly activate pBD-1 and pBD-2 and enhance intestinal epithelial barrier function in porcine jejunal epithelial cell line IPEC-J2. 117
HDACs regulate other factors
In addition to the abovementioned effects on effector cells, HDAC inhibition has also been shown to alleviate IBD through neutrophils, fibroblasts, colonic muscularis externae tissues, colon tissues (Table 1), etc.
Butyrate has been shown to significantly alleviate mouse colitis via the inhibition of neutrophil-associated immune responses and neutrophil extracellular traps formation. 118 A butyrate-releasing derivative, N-(1-carbamoyl-2-phenylethyl) butyramide (FBA), has been reported to inhibit HDAC9, restore H3 histone acetylation, block neutrophils recruitment, and lead to inflammatory remission in colitis. 119 For fibroblasts in CD, the hypoacetylation of H3K27 has been observed during stricture formation in CD, and VPA has been shown to inhibit TGF-β-induced profibrotic metabolites in primary CD fibroblasts, and suppress collagen-I in intestinal fibroblasts through increasing histone acetylation. 120 Nitric oxide (NO) release by S-nitrosoglutathione (GSNO) has been shown to increase the transcription of HDAC3 and suppress H4K12 acetylation at the ICAM-1 promoter to restrict NFκB in the colonic muscularis externae tissues of naïve adult rats, inhibiting ICAM-1 expression in TNBS-induced colitis model. 121 Furthermore, the novel HDAC8 inhibitor SPA3074 has been shown to upregulate suppressor of cytokine signaling 1 (SOCS1) expression in colon tissues, thus strengthening the epithelial barrier and alleviating inflammation in DSS-induced colitis. 122
Conclusion
In the past decade, the landscape of IBD treatment has undergone substantial transformation, largely due to advances in immunomodulatory drug development and the emergence of other novel therapies. However, many more effective and tolerable therapies are needed. Already existed HDACis and new therapy targeting diverse HDACs, as summarized above (Tables 2 and 3), have shown great potential in IBD treatment.
Summary of the mechanisms by which HDACis regulate IBD in different cells and tissues.

BMDM, bone marrow-derived macrophages; HDAC, histone deacetylase; LP, lamina propria; LPS, lipopolysaccharide; NET, neutrophil extracellular trap; PBMC, peripheral blood mononuclear cell; SOCS1, suppressor of cytokine signaling 1; Tregs, regulatory T cells; VDR, vitamin D receptor.
Summary of the mechanisms by which HDACs and histone acetylation regulate IBD in different cells and tissues.

GBP5, guanylate-binding protein 5; GSNO, S-nitrosoglutathione; HDAC, histone deacetylase; IBD, inflammatory bowel disease; NO, nitric oxide.
To date, the FDA has approved five HDACis for use for the treatment of other diseases but not for IBD because of their relatively narrow therapeutic window, short half-life, and adverse events. 50 In a phase I study, panobinostat has been combined with a proteasome, carfilzomib, to treat relapsed/refractory multiple myeloma patients. It was found that the maximum tolerated regime is panobinostat 20 mg with carfilzomib 36 mg/m2, and adverse events include thrombocytopenia, fatigue, and nausea/vomiting. 123 In a phase I/Ib trial, vorinostat has been combined with pembrolizumab, an inhibitor of the programmed cell death protein 1, in the treatment of advanced/metastatic non-small cell lung cancer. No patients showed dose-limiting toxicities but only fatigue and nausea/vomiting as adverse events. The recommended dose is vorinostat 400 mg with perbrolizumab 200 mg in this phase I study. 124 In another phase Ia study, bisthianostat, has been found to be well tolerated in multiple myeloma patients with no grade 3/4 nonhematological adverse events. 125 In previous clinical trials, HDACis are overall safe and the incorporation of HDACis into the therapeutic arsenal for IBD still needs to be explored further, with the aim of increasing patient longevity and improving quality of life.
Future prospects
It is very meaningful and significant for clinical treatment to explore more about the molecular mechanism of HDACs in IBD pathogenesis, including the correlation between HDACs and immunity, connection between HDACs and intestinal microbiota, etc. Furthermore, the ongoing challenge of searching for newer and more effective HDACis with minimal toxicity for IBD underscores the need for further continuous research. What is more, exploring novel therapeutic combinations are urgently needed for IBD treatment optimization, such as combinations of one or two HDACis with other classic treatment for IBD. As research progresses, we anticipate that the targeted application of HDACis will be integrated into the treatment paradigm for IBD, increasing the effectiveness and safety of treatment options for IBD patients.