
Abstract
Pancreatic cancer is a highly aggressive and lethal cancer characterized by high invasiveness, local and extensive dissemination at time of diagnosis and resistance to treatment. Few therapies have shown efficacy in the past and even standard of care therapies yield only modest improvements in the mortality of patients with advanced or metastatic disease. Efforts have been undertaken to study the pancreatic tumor microenvironment and have established its complex and immunosuppressive nature which could explain the high resistance to chemotherapy. Novel therapies targeting the tumor microenvironment with an aim to decrease this resistance, improve immune tolerance and increase the efficacy of the current treatment have shown some promising preliminary results in preclinical and clinical trials. We review the current advances in the field of immunotherapy and their effectiveness as a potential treatment strategy in the pancreatic cancer.
Introduction
Pancreatic cancer is the twelfth most common cancer and is the seventh most common cause of cancer related deaths worldwide with 338,000 new cases and a 5-year prevalence of 4.1 per 100,000 [Ferlay et al. 2015]. In the United States, it is the third leading cause of cancer-related deaths with an estimated incidence of 48,960 cases, an estimated mortality of 40,560 in 2015 and 5-year survival rate of 7.2% [Howlader et al. 2015]. For many years, gemcitabine had been the primary first-line treatment for pancreatic ductal adenocarcinoma (PDAC). The advent of polychemotherapy approaches for locally advanced and metastatic disease using FOLFIRINOX (folinic acid, 5-fluorouracil, irinotecan, oxaliplatin) [Conroy et al. 2011] was able to attain a median survival (MS) of 11.1 months. Von Hoff and colleagues conducted a phase III MPACT trial to study efficacy and safety of nab-paclitaxel and gemcitabine combination versus monotherapy in pancreatic adenocarcinoma. The median overall survival (OS) was 8.5 months in the nab-paclitaxel–gemcitabine group as compared with 6.7 months in the gemcitabine group. The survival rate at 1 year was 35% and 22% and progression-free survival (PFS) was 5.5 and 3.7 months in combination versus monotherapy groups, respectively [Von Hoff et al. 2013]. More recently, Goldstein and colleagues updated results based on long-term follow up and found OS to be 8.7 versus 6.6 months in combination and monotherapy group and found long-term survivors (>3 years) in the combination group (4%) [Goldstein et al. 2015]. There have been many attempts to create other polychemotherapy regimens by combining additional agents with gemcitabine, without significant benefit noted. A randomized phase III trial of pemetrexed in combination with gemcitabine given to 565 locally advanced or metastatic pancreatic cancer demonstrated no significant survival benefits with a MS of 6.5 months and progression-free interval (PFI) of 3.6 months as compared with single-agent gemcitabine [Oettle et al. 2005]. Despite the advances in the treatment for PDAC, patients eventually experience disease progression and cures are rare.
The effect of gemcitabine on the immune system in advanced pancreatic cancer patients has been studied and was not found to be immunosuppressive. Regardless of decreased memory T cell, gemcitabine promotes naïve T-cell activation and may enhance responses to antitumor vaccines and immunotherapy [Plate et al. 2005]. Further effect of gemcitabine proceeded by pemetrexed, an antifolate drug, on innate and adaptive immunity was studied in a phase II study of 15 patients with advanced pancreatic cancer. The study demonstrated MS of 9.5 months and PFI of 4.75 months. Pemetrexed decreased the levels of natural killer (NK) cytolytic units when combined with gemcitabine and initially increased levels of interferon gamma (IFNγ) in NK cells which returned to baseline after the addition of gemcitabine. The combination led to decreased OX40+ activated T cells, helper T cells and memory T cells but levels were increased in subjects with high expression of B7-H3 in tumor tissue. Innate NK cell immunity and FoxP3+ T cells and CD8+ T cells correlated positively with survival and seemed beneficial in patients with pancreatic tumor. Thus, innate and adaptive immunity are important defenses against pancreatic tumors [Davis et al. 2012]. As such, there is a need to further explore novel therapies to improve morbidity and mortality related to PDAC.
Current progress in immunology-based therapy targets the highly heterogeneous pancreatic tumor immunosuppressive microenvironment. Macrophages, myeloid derived suppressor cells and regulatory T cells (Tregs) are the three major leukocyte subtypes found in early pancreatic intraepithelial (PanIN) stage that are involved in immunosuppressive antitumor activity and more advanced PDAC stage is associated with increased number of Tregs and inactivated effector T cells (Teffs) [Clark et al. 2007]. Various animal and human models have suggested that these immunosuppressive cells also increase in the peripheral blood, stroma and accumulate in PDAC tissue hindering the normal effector T-cell proliferation and response [Liyanage et al. 2002; Zhao et al. 2009; Amedei et al. 2013]. An animal model demonstrated that manipulation of these targets may inhibit or restrict tumor growth [Tan et al. 2009]. These observations advocate for the role of immunosuppression in PDAC. In this article, we review recent advances in immunotherapeutics for pancreatic cancer treatment. See Figure 1 for a diagrammatic depiction of mechanisms.

Illustrative image describing immunotherapy mechanisms employed in defense against pancreatic cancer.
Immune checkpoint inhibitors
Tumor peptide antigen presentation by major histocompatibility complex (MHC) to the T-cell receptor (TCR) is helpful in eliciting the antitumor immune response. However, this response is modulated by several regulatory molecules expressed on T cells termed ‘immune checkpoints’. These costimulatory or coinhibitory molecules synergize with TCR signaling to promote or inhibit the immune activity under physiologic and pathologic conditions. Co-regulatory molecules serve as immunotherapeutic targets to modify MHC–TCR signaling to optimize the immune response against cancer cells [Pardoll, 2012; Chen and Flies, 2013].
Anti-CTLA-4
CTLA-4 (CD152) is one of the first immune checkpoint receptors to be targeted clinically [Brunet et al. 1987]. It is involved in regulating early stages of T-cell activation. It belongs to the family of CD28 receptors and has an inducible expression on the surface of CD8+ and CD4+ T cells. CTLA-4 binds to ligands B7-1 (CD80) and B7-2 (CD86) on antigen-presenting cells (APCs), and due to its higher avidity, displace the costimulatory receptor CD28. Thus, by blocking the stimulatory effect of CD28 ligand, it attenuates the immune response by inhibiting activation of Teffs, decreasing interleukin (IL)-2 production and activating Tregs to exert suppression [Blank and Enk, 2015]. Several other extrinsic and intrinsic cell mechanisms are also believed to play a role in CTLA-4 function. These include reverse signaling via B7 that can lead to increased production of indoleamine-2,3-dioxygenase (IDO) decreasing T-lymphocyte activation and proliferation and presence of associated phosphatases SHP-2 and PP2A impairing TCR signaling by dephosphorylation leading to overriding of the TCR-induced stop signal required for stable conjugate formation between T cells and APCs, ultimately leading to reduced cytokine production and proliferation [Schneider et al. 2006; Rudd, 2008]. Collectively, all of these factors are believed to limit the body’s immune response to antigens including self-antigens.
The tumor microenvironment in pancreatic cancer has an increased Tregs/Teffs [Ino et al. 2013] and increased expression of negative costimulatory molecules [Chen and Flies, 2013]. Preclinical murine models demonstrated improved tumor control and its shrinkage after CTLA-4 blockade [Leach et al. 1996; Van Elsas et al. 1999; Peggs et al. 2009] which led to the clinical development of the anti-CTLA-4 agent, ipilimumab [Hodi et al. 2010]. Improvement of OS by 4 months (10 versus 6.4 months) led to US Food and Drug Administration (FDA) approval of ipilimumab in metastatic melanoma.
Ipilimumab in pancreatic cancer
Ipilimumab is a fully humanized IgG1 monoclonal antibody (mAb) that antagonizes CTLA-4. A phase II trial [Royal et al. 2010] evaluated the efficacy of ipilimumab for advanced pancreatic cancer in 27 subjects with good performance status. An intravenous dose of 3.0 mg/kg every 3 weeks, four doses per course for a maximum of two courses were given. There were no responders by Response Evaluation Criteria in Solid Tumors (RECIST) criteria but one subject experienced a delayed tumor regression after an initial assessment of progressive disease. Given that RECIST or World Health Organization (WHO) criteria may not provide complete assessment of antitumor response with immunotherapeutics or other such novel agents, a systematic criterion classified as immune-related response criteria was defined to capture and to better interpret any additional atypical responses observed with immune therapy [Wolchok et al. 2009].
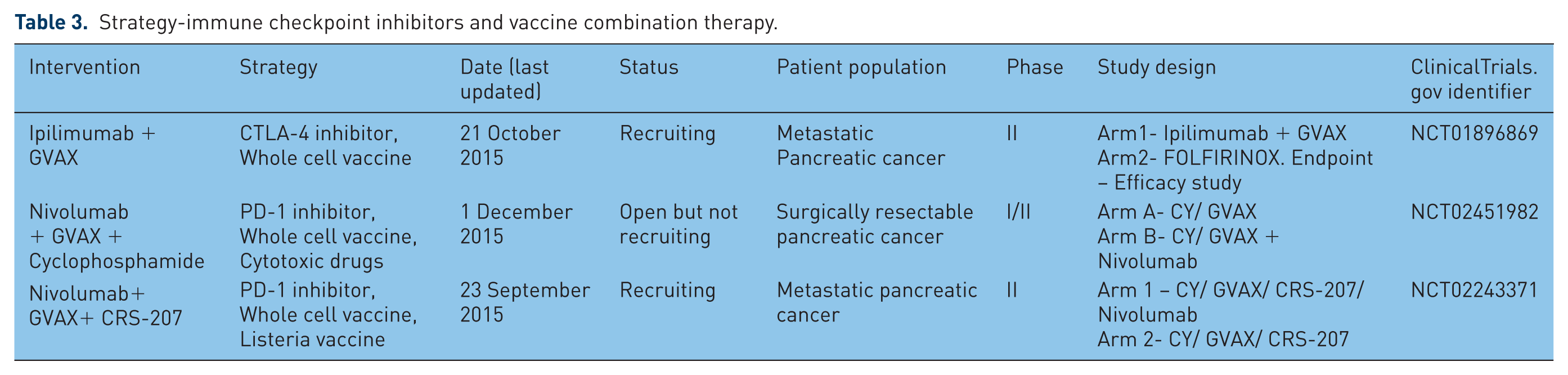
A phase Ib trial [ClinicalTrials.gov identifier: NCT00836407] was performed to determine clinical safety, survival rates and T-cell responses to ipilimumab 10 mg/kg alone (arm 1) and in combination with granulocyte macrophage colony-stimulating factor (GM-CSF) vaccine GVAX (arm 2) in patients with previously treated PDAC. Two patients in arm 1 and three patients in arm 2 demonstrated stable disease, and seven patients in arm 2 noted a decline in CA19-9. The combination arm demonstrated an improved median OS (3.6 versus 5.7 months) and 1-year OS (7% versus 27%) [Le et al. 2013]. Additional investigation into optimal use, clinical safety and efficacy of ipilimumab alone or in combination has led to several ongoing clinical trials as documented in Tables 1–5.
Strategy-immune checkpoint inhibitor monotherapy.

Strategy-dual checkpoint inhibitor combinations.

Strategy-immune checkpoint inhibitors and vaccine combination therapy.
Strategy-immune checkpoint inhibitors with chemotherapy.

Strategy-immune checkpoint inhibitors and tyrosine kinase inhibitors combination therapy.

Tremelimumab in pancreatic cancer
Tremelimumab is a human IgG2 mAb that blocks CTLA-4 [Ribas et al. 2007]. Phase II/III clinical trials in patients with advanced melanoma treated at a dosage of 15 mg/kg every 3 months failed to yield any significant survival benefit [Kirkwood et al. 2010; Ribas et al. 2013]. A phase II single-arm study in patients with malignant mesothelioma yielded a PFS of 6.2 months and a median OS of 10.7 months [Calabrò et al. 2013]. Clinical trials for tremelimumab in pancreatic cancer are underway.
Anti-PD1 and anti-PD-L1
Programmed cell death protein-1 (PD-1)/CD279 is a coinhibitory molecule of the CD28 family. It is expressed predominantly by activated CD4+ and CD8+ T cells, Tregs [Francisco et al. 2010] and is also found on activated B cells [Pedoeem et al. 2014], NK cells, monocytes and certain dendritic cells (DCs). Its binds to the PD-L1 (B7-H1) and PD-L2 (B7-DC) ligands of family B7. PD-L1 is constitutively expressed on several solid tumor cells and tumor-infiltrating lymphocytes (TILs) [Dong et al. 2002] while PD-L2 is expressed on DCs, macrophages, B-cell subsets [Pedoeem et al. 2014] and is upregulated in lymphomas and hematologic malignancies [Rosenwald et al. 2003]. PD-1 engagement with its ligands controls peripheral T-cell tolerance. PD-L1 interaction with PD-1 and B7-1 (CD80) results in negative signals leading to decreased T-cell proliferation, cytokine release and cytolytic activity of PD-1+ T cells. Chronic antigen exposure such as cancer and viral infection, can lead to persistent PD-1 expression inducing a state of exhaustion among antigen-specific T cells and helping the tumor cells escape from host immune attack. Blockade of these targets reverses the tumor protective effects and may provide an effective approach for tumor immunotherapy [Iwai et al. 2002; Okudaira et al. 2009]. Increased expression [Razzaque et al. 2013] of PD-L1 and PD-L2 significantly correlates with worse prognosis, higher grade and is inversely correlated with TILs in pancreatic cancer [Nomi et al. 2007; Geng et al. 2008]. Furthermore, its role in pancreatic adenocarcinoma is defined, given that knocking down of B7-H1 (PD-L1) inhibits PDAC proliferation [Song et al. 2014].
Nivolumab and pembrolizumab, examples of anti-PD1 therapeutic agents, have demonstrated positive results with in various studies involving malignant melanoma [Mahoney et al. 2015], renal cell carcinoma [Weinstock and Mcdermott, 2015] along with preliminary efficacy in a wide array of other tumors. Pembrolizumab has been approved by US FDA for malignant melanoma previously treated with ipilimumab [Robert et al. 2014]. The first PD-L1 antibody to show an objective response in a phase I study including patients with solid tumors was BMS-956559 [Brahmer et al. 2012], but no responses in patients with pancreatic and colorectal cancer were observed. Various PD-1 and PD-L1 inhibitors are presently in clinical trials for pancreatic malignancies.
A broad array of clinical trials in pancreatic cancer alone or advanced solid tumors including pancreatic cancer have been completed or ongoing using immune checkpoint monotherapy (Table 1), dual checkpoint combination therapy (Table 2), immune checkpoint inhibitor and vaccine combinations (Table 3), immune checkpoint inhibitors with cytotoxic chemotherapy (Table 4), immune checkpoint inhibitors with kinase inhibitors (Table 5) and immune checkpoint inhibitor combinations with other agents (Table 6).
Strategy-other therapies in combination with immune checkpoint inhibitors.

Therapeutic vaccine immunotherapy
The principle of using vaccine immunotherapy in cancer treatment is similar to passive immunity against an infectious process. These are biologically designed to mount an immune response against a tumor by administering tumor-specific antigens. They include whole-cell vaccines, DC vaccines, DNA vaccines and peptide vaccines.
Whole-cell vaccines
Whole-cell recombinant vaccines utilize the tumor cells which have a wide range of antigens and express epitopes for CD8+ and CD4+ T cells. Allogeneic cell lines may be used instead of autologous cell lines in pancreatic cancer when it is difficult to harvest these tumor cells due to limited number of surgical candidates, long culture times and risk of contamination [Koido et al. 2005].
Algenpantucel-L
Algenpantucel-L is a hyperacute vaccine that has allogenic irradiated pancreatic cancer cells expressing the enzyme alpha-1,3-galactosyl transferase (αGT) which is required for the synthesis of alpha-galactosyl (αGal) epitopes (Table 7). Humans lack the presence of αGal epitopes but have anti-αGal antibodies [Galili et al. 1985] and these are continuously produced in response to enterobacteria with no immune tolerance to it [Galili et al. 1988]. The hyperacute technology exploits this naturally acquired immunity against αGal labeled tumor cells leading to enhanced antitumor response [Mandell et al. 2009]. Effective tumor responses in animal models [Gorelik et al. 1995; Rossi et al. 2005] led to subsequent human clinical trials. Hardacre and colleagues [Hardacre et al. 2012], presented results of an open-label, single-arm phase II study of algenpantucel-L with gemcitabine and 5-fluorouracil for 70 resected pancreatic cancer patients. One-year disease-free survival (DFS) of 63% and OS of 86% was achieved as compared with historical controls (~45% and 65%). Current ongoing phase III clinical trials are the Immunotherapy for Pancreatic Resectable Cancer Survival Study (IMPRESS) and the Pancreatic Immunotherapy with algenpantucel-L for Locally Advanced non-Resectable (PILLAR) trial.
Algenpantucel-L in clinical trials.

GM-CSF vaccine
GVAX is an allogenic irradiated whole-cell tumor vaccine transfected with GM-CSF gene and delivering tumor-associated antigens (TAAs) at the same time (Table 8). GM-CSF is a potent cytokine released by macrophages, T cells, mast cells, NK cells that function to stimulate stem cells to produce granulocytes and monocytes, promoting cytolytic activity against tumor cells [Thomas et al. 2004] and also causing influx of bone-marrow derived DCs that process and present TAAs delivered by the vaccine [Huang et al. 1994]. A phase I [Jaffee et al. 2001] clinical trial of GVAX in resectable pancreatic cancer given before and after chemoradiation therapy, assessed 14 patients and showed a mean DFS of 13 months. Three patients who demonstrated delayed-type hypersensitivity (DTH) were disease free from 25 to 30 months. A dose of 50 × 107 vaccine cells was identified as safe with limited side effects and associated with induction of antitumor immune response. These favorable results resulted in phase II trials of GVAX in combination with cyclophosphamide (CY) [Laheru et al. 2008] in metastatic pancreatic cancer patients who failed gemcitabine therapy. Despite no significant statistical difference of OS and 1-year survival between two cohorts, this study suggested enhanced antitumor activity in combination cohort where CY is given prior to immunotherapy based on a higher rate of induction of mesothelin-specific T-cell responses that could be associated with longer PFS and OS. Another phase II study of 60 patients with resected PDAC received GVAX integrated with boosters and 5-FU-based chemoradiation demonstrated a median DFS of 17.3 months with MS of 24.8 months. It also demonstrated a correlation between induction of mesothelin-specific T-cell responses with improved OS proposing a prognostic role [Lutz et al. 2011]. Additional markers that may be associated with prolonged survival in patients treated with GVAX and/or ipilimumab are induction of thyroglobulin antibodies due to thyroid autoimmunity [De Remigis et al. 2015].
Clinical trials that used combination of whole cell vaccine GVAX and chemotherapy.

Peptide vaccines
Peptide-based vaccines constitute tumor antigenic fragments (epitopes) which elicit a tumor-specific response by stimulating T cells. These vaccines are safe, simple and economically beneficial. However, their minimal immunogenic potential, impaired antigen presentation [Yanagimoto et al. 2005] and defective immune response in patients with advanced cancer have limited their usefulness [Gaudernack, 2006].
Ras vaccine
K-Ras is mutated in almost 90% of pancreatic cancers [Hruban et al. 1993] and is presented as a foreign antigen by MHC class I and II to CD4+ and CD8+ T cells [Abrams et al. 1997; Gjertsen et al. 1997]. Therefore, K-Ras mutated gene product can serve as a focus for therapeutic peptide vaccine inducing anti-Ras immunity in patients. Initial studies testing Ras vaccine in humans [Gjertsen et al. 1995] demonstrated the safety and induced transient T-cell tumor response against mutations in tumor cell, albeit with no clinical responses. Failure to manifest clinically significant immunity was attributed to the inability to elicit the imperative immune response to these antigens. Eventually in an attempt to enhance the immune response, a phase I/II trial combining synthetic mutant Ras peptides with GM-CSF as an adjuvant was given to 48 patients (10 surgically resected and 38 with advance disease) of pancreatic adenocarcinoma. Peptide-specific immunity was induced in 58% of patients and the responders showed an improved survival over nonresponders (MS 148 days versus 61 days, respectively) and 9 of 10 resected PDAC patients showed stability with an OS of 25.6 months (range 10–39 months) and 11 of 34 unresectable patients had a stable disease lasting an average of 10.2 months (range 3–28 months) [Gjertsen et al. 2001].
Adjuvant use of Ras vaccine in a phase II clinical trial [Toubaji et al. 2008] in patients with surgically resected (five pancreatic and seven colorectal cancer patients) with no evidence of disease at enrollment demonstrated a positive immune response with an increase of IFNγ m-RNA copy numbers in 5/11 patients. Pancreatic cancer patients had a mean DFS of 35.2+ months and a mean OS of 44.4+ months providing evidence for adjuvant early use of mutant Ras peptide immunotherapy. Subsequently [Wedén et al. 2011], a study using a long synthetic mutant Ras peptide vaccine described a 10-year follow up of 23 patients from previous phase I/II clinical trials [Carbone et al. 2005; Toubaji et al. 2008] to assess their long-term immunological T-cell reactivity and survival. A total of 17 of 20 (85%) were immune responders with a MS of 28 months. The 5-year survival was 22% and 28% for all and immune responders respectively and a striking 10-year survival of 20% (4/20 evaluable) versus 0% in comparable nonvaccinated cohort, was observed. Recently, another study evaluated 24 patients with resected pancreatic cancer with K-Ras mutation that were vaccinated with peptide vaccine on day 7 of a 10-day course of GM-CSF. A total 25% of patients (9/24) were immune responders, three of them displayed nonspecific DTH and one patient had a K-Ras mutation-specific DTH. Median recurrence-free survival time was 8.6 months and median OS was 20.3 months [Abou-Alfa et al. 2011]. These studies demonstrate promising results and suggest larger controlled studies depicting the role of K-Ras vaccine as an adjuvant immunotherapy for PDAC are needed.
Another strategy applied to activate immune system against the mutated Ras peptide is by using nonpathogenic yeast as a vehicle to carry antigens and present them to DCs. DCs treated with yeast and cocultured with T cells had a lower expression of FoxP3 cells and increase in the ratio of CD4+CD25+ activated T cells to Tregs and production of Th1-related cytokines and IL-6 [Cereda et al. 2011]. GI-4000 is a product series of four tarmogens; each tarmogen is a different heat inactivated yeast Saccharomyces cerevisiae expressing combination of common Ras mutations found in human cancers. Clinical trials studying the K-Ras peptide vaccine alone or as combination therapy in pancreatic cancer are shown in Table 9.
Ras vaccine in clinical trials.

Telomerase peptide vaccine
Telomerase is a ribonucleoprotein enzyme found in almost 90% cancer cells [Vasef et al. 1999] including pancreatic cancer cells [Suehara et al. 1997]. The induction of human telomerase reverse transcriptase (hTERT), a telomerase catalytic subunit parallels the increased expression of the enzyme [Counter et al. 1998]. Acknowledging its role in tumorigenesis, telomerase tumor antigen served as a target for the development of peptide-based vaccine, GV1001. It is a 16-aa hTERT peptide that binds to MHC activating hTERT-specific T-cell responses [Su et al. 2005]. GV1001 was studied in a phase I/II trial by Bernhardt and colleagues in 48 patients with unresectable pancreatic cancer. The GV1001 vaccine was given in three dose levels (100, 300, 1000 nmol) along with GM-CSF for a period of 10 weeks with monthly boosters. Immune response (positive DTH test or the presence of GV1001-specific T cells in peripheral blood after vaccination) was observed in 24 of 38 evaluable patients with the highest immune response (75%) in the intermediate dosage group. The MS for the low-, intermediate- and high-dose groups were 4.0, 8.6 and 5.1 months, respectively. As such, prolonged immunologic response correlated with prolonged survival in the study [Bernhardt et al. 2006].
The two phase III trials testing PrimoVax (GV1001 and GVAX) [Buanes et al. 2009] and TeloVac (GV1001) [Middleton et al. 2013] administered with subsequent and concurrent chemotherapy were unable to simulate similar affirmative results and showed no significant survival benefits in patients with metastatic pancreatic cancer. A currently ongoing clinical trial [ClinicalTrials.gov identifier: NCT01342224] is testing GV1001 with GM-CSF in patients with locally advanced pancreatic cancer given in combination with gemcitabine chemotherapy and tadalafil (PDE5 inhibitor) followed by concurrent radiation therapy.
Mucin-1 vaccine
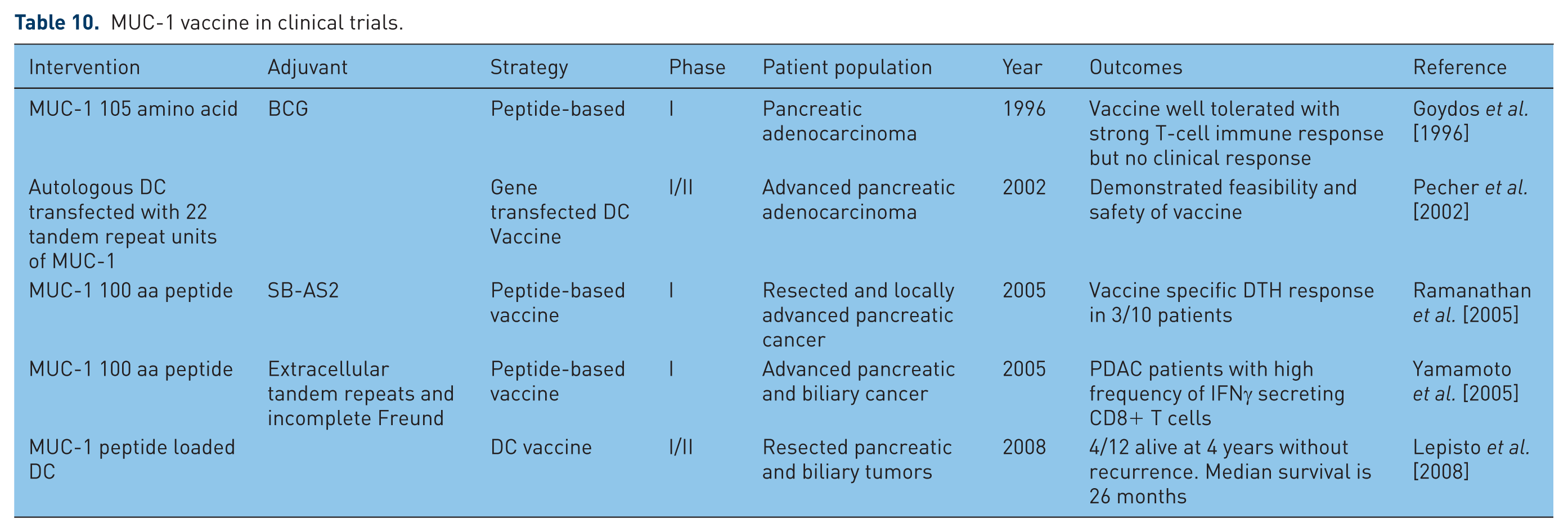
Mucin 1 (MUC-1) is type I transmembrane protein constituting a large extracellular domain with extensive O-linked glycosylation and a small cytoplasmic domain in close contact with the nucleus and plays a role in normal cell signal transduction as well as oncogenic signaling to increase invasion, angiogenesis and metastasis [Behrens et al. 2010]. MUC-1 expression is increased in tumor cells including pancreatic tumors [Burdick et al. 1997; Qu et al. 2004] with a loss of polarity and hypoglycosylation. The overexpression and aberrant glycosylation makes MUC-1 highly immunogenic and, thus, a target for cancer immunotherapy [Besmer et al. 2011]. MUC-1 vaccine has been tested in different forms such as peptide vaccine, peptide transfected DC, DCs transfected with c-DNA22 tandem repeats units of MUC-1, as well as adoptive transfer immunotherapy. The phase I clinical trials by Goydos and colleagues [Goydos et al. 1996], Ramanathan and colleagues [Ramanathan et al. 2005] and most recently Yamamoto [Yamamoto et al. 2005] used peptide-based vaccines. Pecher and colleagues [Pecher et al. 2002] tested three different types of formulation of mucin vaccine namely: (1) MUC-1 peptide admixed with murine GM-CSF as an adjuvant; (2) MUC-1 peptide admixed with adjuvant SB-AS2; and (3) MUC-1 peptide-pulsed DCs. Only MUC-1 peptide loaded DC vaccine was able to elicit tumor rejection and response correlated with induction of MUC-1-specific T-cell response in wild and transgenic mice. This was further tested by Lepisto and colleagues [Lepisto et al. 2008].
These studies (Table 10) that define the effect of MUC-1 immunotherapy on pancreatic cancer reveal the safety of the vaccine and has led to increased survival in few vaccinated subjects. MUC-4, MUC-16 are the other potential targets that play a role in PDAC development and also lead to resistance of chemotherapeutic drugs [Bafna et al. 2009; Skrypek et al. 2013] Overcoming this resistance by combining chemotherapeutics with MUC based therapy may improve the clinical outcomes of patients with pancreatic cancer.
MUC-1 vaccine in clinical trials.
Anti-VEGFR vaccine
Vascular endothelial growth factor receptor 2 (VEGFR2) is upregulated and essential in tumor angiogenesis. A phase I trial combining VEGFR2-169 with gemcitabine was conducted in advanced pancreatic cancer patients. A total of 15/18 patients developed immunological response (cytotoxic T lymphocyte and Treg response). A total of 15/18 (83%) developed immunological reactions at the injection sites. Specific cytotoxic T lymphocytes (CTLs) reacting to the VEGFR2-169 peptide were induced in 11 (61%) of the 18 patients. The disease control rate was 67%, and median OS time was 8.7 months [Miyazawa et al. 2010]. A phase I study with pancreatic cancer patients testing gemcitabine with antiangiogenic peptide vaccine (VEGFR2-169) NCT00622622 and a phase I/II study [ClinicalTrials.gov identifier: NCT00655785] with VEGFR2-169 and VEGF1-1084 has been completed.
Survivin-based vaccine
Survivin is one of the cancer-specific proteins overexpressed in tumor cells and human fetal tissues, but hardly detectable in normal human tissues. It promotes angiogenesis, inhibits apoptosis and enhances proliferation [Ryan et al. 2009]. Expression of survivin is increased on pancreatic adenocarcinoma tissue as well as in serum [Dong et al. 2015] and is associated with poor prognosis and resistance to chemotherapy and radiation [Kami et al. 2004]. A 77-year-old patient suffering from pancreatic cancer with liver metastasis refractory to gemcitabine therapy received a survivin-based vaccine (consisting of 100 μg of a modified HLA-A2 restricted surviving (96–104) epitope in Montanide(R)) and achieved a complete remission of liver metastasis at 8 months but with recurrence of disease after being weaned from the vaccine [Wobser et al. 2006]. This effect was further tested in a study [Ishizaki et al. 2011] on the Pan02 mouse pancreatic adenocarcinoma cell line that combined gemcitabine and modified vaccinia Ankara (MVA) expressing murine survivin. This study resulted in significant tumor regression and prolonged survival. A total of 50% of the mice in the MVA-survivin + gemcitabine group were alive at 50 days, whereas all other animals in MVA-survivin and MVA-control + gemcitabine groups expired by 38 days post-tumor induction. These results observed with combined immunotherapy were primarily dependent on generation of CD8+ T cells. Gemcitabine and survivin act in synergy creating an antitumor environment by generating effector cells against self-tumor antigen and apoptosis of tumor cells that helps to reduce tolerance against survivin and prolong the survival in combination therapy. The data from the study suggests combination therapy may find its potential for clinical benefit in pancreatic cancer.
Antigastrin vaccine
Gastrin-17 is a growth factor for pancreatic and other gastrointestinal malignancies. Antigastrin-17 vaccine (G17DT) has a gastrin immunogen with diphtheria toxoid (DT) carrier protein. The first phase II study testing G17DT recruited 30 patients with advanced pancreatic cancer, out of which 20 patients produced an antibody response. The MS for immune responders was 217 days and 121 days for nonresponders (p = 0.0023) [Brett et al. 2002]. A randomized clinical trial of 154 patients with advanced pancreatic cancer was well tolerated (2/79 (2.5%) G17DT patients versus 0/74 (0%) placebo patients developed sterile abscess at the injection site). The MS in the G17DT group was 138 days versus 78 days for the control group [Gilliam et al. 2004]. Subsequently, a randomized, multicenter, double-blind, placebo-controlled study of 154 patients with advanced pancreatic cancer gave MS of 151 days as compared with 82 days with placebo [Gilliam et al. 2012]. Another multicenter study evaluated the role of G17DT + gemcitabine versus placebo + gemcitabine in 394 patients with locally advanced, recurrent or metastatic adenocarcinoma of pancreas. The MS was 5.8 months in G17DT + gemcitabine group which was lower than the placebo + gemcitabine (6.6 months) and same PFS of 3.9 months [Shapiro et al. 2005]. A prospective randomized phase III study testing G17DT for the treatment of advanced pancreatic cancer [ClinicalTrials.gov identifier: NCT02118077] was recently completed and results are awaited.
Heat shock protein–peptide complex
Heat shock proteins act as peptide carriers for stabilization and delivery. Overexpression of HSP90 is observed in pancreatic carcinoma and HSP90 β is reported to inhibit apoptosis [Ogata et al. 2000]. A phase I pilot study of autologous heat shock protein vaccine HSPPC-96 in patients with resected pancreatic adenocarcinoma to determine tolerance and immune as well as clinical responses. A total of 3/10 patients were alive at 2.6, 2.7 and 5 years follow up [Maki et al. 2007]. A phase I clinical trial of immunotherapy with autologous tumor-derived gp96 heat shock protein-peptide complex in patients with resected pancreatic adenocarcinoma [ClinicalTrials.gov identifier: NCT00003025] has been completed and results are awaited.
Dendritic cell vaccine
DCs are the most potent APCs which function to process and present antigens with MHC to activate cytolytic and regulatory T-cell response [Koski et al. 2008]. These characteristics are being employed in development of DC vaccines to mount an antitumor response (Table 11). DC vaccines are designed to present TAAs like peptides, mRNA or cDNA or by fused autologous DC with tumor cells and means by which they are presented including: (a) nontargeted vaccine which consists of tumor-specific peptides or nucleic acids captured in vivo; (b) vaccines with ex vivo generated DCs loaded with antigens; (c) vaccines with tumor antigens coupled to anti-DC-antibodies to mount in vivo response [Palucka and Banchereau, 2013]. Intratumoral response with tumor RNA-pulsed DC was studied in C57BL/6 murine pancreatic tumor model inducing a specific T lymphocyte antitumor effect [Schmidt et al. 2003].
Studies using dendritic cell (DC) vaccines.

Further clinical trials to determine the effectiveness of CEA RNA-pulsed DC vaccine alone [ClinicalTrials.gov identifier: NCT00004604] and in combination with chemotherapy gemcitabine [ClinicalTrials.gov identifier: NCT00547144] and chemokine modulatory regimen (CKM) [ClinicalTrials.gov identifier: NCT02151448] are in process.
Recombinant bacterial vaccine
Bacterial agents such as Listeria monocytogenes (LM) and Salmonella enterica typhimurium act as delivery systems for TAAs because of their ability to survive intracellularly [Paterson et al. 2010]. Listeria is a Gram-negative intracellular facultative bacterium which is actively phagocytosed by macrophages or other APCs. Once inside the phagosome it secretes Listeriolysin O (LLO) and phospholipase C which helps it to modify the phagosomes to larger compartment called spacious Listeria-associated phagosomes (SLAPS) [Birmingham et al. 2008] promoting its replication and release from the phagolysosome into the cytoplasm. LM-based immunotherapy utilizes live attenuated LM bacteria to act as a vaccine vector and deliver the TAAs to the APCs and hence eliciting an antitumor immune response.
Preclinical studies have shown the antitumor effect of the vaccine. A study in FVB/N mouse fusing LLO with HER-2/neu antigen delivered by LM showed the increased immunogenicity of self-antigens and provided tumor regression [Singh et al. 2005]. CRS-207 is a live attenuated vaccine of LM bacterium has two gene deletions that encode for virulence and is modified to express mesothelin. A phase I clinical trial [Le et al. 2012] tested ANZ-100 (live attenuated LM strain) and CRS-207 in advanced mesothelin expressing cancers with liver metastasis. CRS-207 induced both LLO and mesothelin-specific T-cell responses and chemokine induction were present at all doses while ANZ-100 induction of chemokines was dose dependent. While the study was statistically unable to show survival benefit, it revealed that 37% of the population survived for more than 15 months with 3 PDAC long-term survivors with minimal adverse events. A more recently completed randomized phase II study assessed safety and survival of CRS-207 with GVAX and low-dose CY for 90 metastatic pancreatic cancer patients [Le et al. 2015]. GVAX/CY inhibits Tregs and had shown earlier preclinical synergy with the vaccine that helps to induce innate and adaptive immunity. OS was 9.7 months with an improvement of 5.6% (2.2 months) as compared with first-line therapy gemcitabine plus nab-paclitaxel. The longer OS was associated with enhanced mesothelin-specific CD8 T-cell response. An ongoing phase II clinical trial is testing the safety and efficacy of GVAX (with CY) and CRS-207 compared with CRS-207 alone in adults with previously treated metastatic pancreatic adenocarcinoma [ClinicalTrials.gov identifier: NCT02004262].
Adoptive T-cell immunotherapy
Adoptive T-cell immunotherapy (ACT) employs the technique of genetically modifying the T cells and infusing them into tumor tissue to increase the immune response against the tumor. Autologous TILs are engineered to equip them with activity against TAAs using modified TCRs or Chimeric Antigen Receptors (CARs). CARs are recombinant receptors with an intracellular signaling portion constituting T-cell receptor–CD3-ζ complex and an extracellular single chain variable fragment of mAb [Gross et al. 1989; Khan et al. 2014]. The first generation CARs have FcRγ or CD3ζ chains and second-generation CARs incorporates additional cytoplasmic costimulatory domains such as CD28 and CD137 and third-generation CARs comprise three signaling domains. This helps T cells to target tumor cells in a HLA-independent manner to a wide range of surface TAAs [Kershaw et al. 1996].
A preclinical study on murine model expressing anti-CEA CAR T cells were able to reduce the size of pancreatic tumors below the limit of detection and long-term tumor eradication in 67% of mice [Chmielewski et al. 2012]. Intratumoral injection of T cells expressing CAR against Her2/neu or CD24 completely eliminated the tumor and more than 50% of animals appeared to be disease-free more than 2 months later [Maliar et al. 2012]. Similarly other studies with T cells expressing MUC-1 CAR T cells plus IL-4 receptor α [Wilkie et al. 2010] and mesothelin CAR T cells [Carpenito et al. 2009, Zhao et al. 2010] resulted in tumor regression in large, pre-established tumors in mice.
MORAb-009 is a chimeric IgG1 antibody targeting tumor-associated mesothelin overexpressed on pancreatic, ovarian, lung and colorectal carcinoma. A preclinical evaluation of MORAb-900 found the increased effectiveness that led to reduction in tumor growth of mesothelin positive tumors when combined with chemotherapy [Hassan et al. 2007]. This was tested further in a phase I clinical study [Hassan et al. 2010] in 24 mesothelin positive patients including 7 pancreatic cancer patients which showed it was safe and well tolerated at a dose of 200 mg/m2. A total of 11 of previously treated patients in this group had stable disease. Recently reported results of randomized phase II clinical trial [ClinicalTrials.gov identifier: NCT00570713] of MORAb-009 in combination with gemcitabine or placebo in 155 advanced pancreatic cancer subjects failed to suggest any significant benefit of MORAb-900 therapy. The OS of MORAb arm was found to be 6.5 months (4.5–8.10) versus 6.9 months (5.4–8.8) in the placebo arm and PFS of 3.4 months (1.9–4.7) and 3.5 (2.8–4.9), respectively. A total of 19.2% of the patients had progressive disease in the MORAb-009 arm as compared with 23.4% patients in the placebo arm.
Despite the results of the above-mentioned trials, majority of patients has failed to respond well to ACT. Tumor-induced immunosuppression, i.e. upregulation of inhibitory receptors and ligands of activated T cells have shown to contribute to failure of T-cell therapy [Abate-Daga et al. 2013]. To overcome these inhibitory signals, Kobold and colleagues developed a new fusion PD-1 – CD28 receptor hypothesizing that CD28 costimulation may overcome peripheral tolerance [Kobold et al. 2015]. Treatment of mice with Panc02 pancreatic carcinoma cell line expressing ovalbumin (Panc-OVA) with PD-1–CD28 fusion receptor led to complete regression of tumor and had a memory response in 9 out of 11 mice. It warrants further development of the strategy to enhance efficacy of ACT in cancer. Another approach tested preclinically to improve the activity of antitumor CAR T cells is through increased constitutive expression of CD40L. T cells demonstrated increased proliferation and secretion of proinflammatory Th1 cytokines and increased CD19-specific CAR/CD40L T cells cytotoxicity against tumor cells. It also augmented immunogenicity of CD40+ tumor cells by upregulated expression of various costimulatory, adhesion molecules thereby counteracting the tumor cells ability for immune evasion. These novel approaches should be tested in additional studies with the goal of ascertaining their usefulness in a clinical setting. Completed and ongoing studies with adoptive T-cell therapy in pancreatic cancer alone or in solid tumor cancer groups including pancreatic cancer are depicted in Table 12.
Adoptive T-cell therapy in clinical trials.

Immune modulating agents
CD40 agonist antibodies
CD40 is a cell surface receptor that belongs to the family of tumor necrosis factor receptor and is expressed on various APCs including DCs, monocytes, macrophages, mast cells etc. and its ligand CD40L (CD154) is a type II transmembrane protein predominantly expressed on activated T cells. CD40–CD40L interactions provide proliferative signals to promote differentiation of CD4+ and CD8+ T cells and hence may play a role in upregulation of immunity [Grewal and Flavell, 1998; Van Kooten and Banchereau, 2000]. As described earlier, pancreatic cancer leads to immune suppression within the tumor environment by inhibiting costimulatory molecules and also via upregulation of inhibitory molecules on inactivated T cells. Activation of CD40 has shown to restrict tumor growth in vitro and in vivo in many preclinical studies [Funakoshi et al. 1994; Eliopoulos et al. 2000; Tong et al. 2001]. Clinical activity of CD40 molecule was studied by giving CP-870,893, a fully human and selective CD40 agonist mAb in 29 patients with advanced solid tumors. A partial response was observed in four patients with metastatic melanoma and dose-related upregulation of costimulatory molecules on B cells in peripheral blood was also noted [Vonderheide et al. 2007].
Pancreatic cancer tissues have a higher expression of CD40 as compared to adjacent normal tissues. In a study by He and colleagues [He et al. 2012] 69% patients had a positive expression of CD40 and it correlated with higher TNM staging and lymph node metastasis. Also there were increased serum sCD40L levels compared with the healthy subjects. Recombinant CD40L leading to activation of CD40 significantly inhibited the proliferation of pancreatic cancer cell lines and induction of apoptosis. In a phase I study patients with advanced solid tumors were infused weekly doses of CP-870,893 which were well tolerated but there was little clinical activity [Rüter et al. 2010]. A combination of CD40 agonist antibody was tested in combination with gemcitabine in genetically engineered mouse model of PDAC. There was a depletion of CD40+ macrophages in stroma which infiltrated the tumor and became tumoricidal suggesting the CD40+ T-cell-independent mechanism of tumor regression [Beatty et al. 2011]. This was subsequently tested in clinical study. Beatty and colleagues investigated maximum tolerated dose, safety and antitumor activity of CP-870,893 with gemcitabine combination in 22 patients with advanced PDAC. Using RECIST criteria, four patients had a partial response, 11 with stable disease with PET demonstrating more than 25% decrease in FDG uptake within primary pancreatic lesions. One patient with partial response also had a complete regression of 7.6 cm liver mass and 47% regression of a second liver metastasis. Increase in B-cell expansion of costimulatory molecules was seen along with markedly increased inflammatory cytokines. PFS of 5.6 months and OS of 7.4 months was promising. Improved OS correlated with a decrease in FDG uptake in hepatic lesions suggesting the early role of FDG-PET/CT imaging [Beatty et al. 2013]. Table 13 summarizes completed and ongoing studies with CD40 agonist antibodies.
Studies using CD40 agonist antibody.

(C-C motif) receptor 2 inhibitor
CCR2 is a chemokine receptor which mediates chemotaxis of immune cells and is expressed on monocytes and macrophages. Chronic tumor infiltrating cells might play a role in tumor progression, metastasis and resistance to chemotherapy. Targeting tumor-associated macrophages (TAMs) by inhibiting CCR2 would decrease the immune cells in tumor environment and increases efficacy of chemotherapy [Mitchem et al. 2013]. PF-04136309 is a novel CCR2 inhibitor with preclinical antitumor activity in PDAC through depleting TAMs and inflammatory monocytes. A phase Ib study of PF-04136309 given with FOLFIRINOX in 41 patients was conducted. A total of 12 of 23 evaluable patients (52.2%) had partial response by RECIST and 11 (47.8%) had stable disease. Curative resection was achieved in four out of five patients with borderline resectable and two with locally advanced disease [Wang-Gillam et al. 2015]. The data thus far are promising and need to be further validated.
Conclusion
Immunotherapy is a promising emerging treatment in cancer care. Manipulation of the immune targets along with traditional chemotherapy displays a synergy in antitumor response suggesting a probable role for immunotherapy as an adjuvant or in combination with radiochemotherapy. Furthermore, combining different immunotherapeutic strategies has shown some favorable outcomes with significant clinical benefits. Additional larger clinical trials testing the complex of immune checkpoint inhibitors with use of one or more types of vaccines and newer personalized therapies are warranted. Pancreatic cancer is a systemic disease which remains highly lethal even when resected. Therefore, early administration of effective combination therapy including immunotherapy may prove beneficial and needs further evaluation.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there is no conflict of interest.