
Abstract
Hermansky–Pudlak syndrome (HPS) is a rare autosomal recessive disorder caused by defects in lysosome-related organelles (LROs). Eleven HPS subtypes have been identified, each associated with mutations in distinct genes, with HPS-4 being among the rarer forms. Here, we present a 38-year-old Chinese woman with oculocutaneous albinism (OCA), bleeding tendency, and progressive pulmonary fibrosis, ultimately diagnosed with HPS through platelet transmission electron microscopy. The patient received nintedanib, an antifibrotic agent, which appeared to stabilize pulmonary function for approximately 18 months. Genetic testing revealed a novel homozygous splice site variant in HPS4 (NM_022081.6 c.1713+1delG), predicted to result in splice disruption and nonsense-mediated mRNA decay. Based on in silico analysis, the variant was considered pathogenic. To contextualize this case, we reviewed all published HPS4-related cases and identified 51 reported patients carrying 37 unique mutations, including 16 nonsense, 8 frameshift, 7 splice-site, 5 missense, and 1 gross insertion variant. Our findings broaden the mutational spectrum of HPS4 and support the utility of nintedanib in managing HPS-associated interstitial lung disease. It also underscores the potential for pharmacologic intervention to alter the disease trajectory in HPS, particularly in resource-limited settings where lung transplantation is not widely accessible.
Introduction
Hermansky–Pudlak syndrome (HPS) is a rare multisystem disorder owing to dysfunction of cell type specific lysosome-related organelles (LROs), characterized by oculocutaneous albinism (OCA) or ocular albinism, bleeding tendency, and/or other complications such as pulmonary fibrosis, colitis, immunodeficiency, and neuropsychological disorders. 1 It is an autosomal recessive hereditary disease, with an estimate prevalence of 1/350,000–1/1,000,000 worldwide, but is more common in a section of Puerto Rico. 2
HPS is a rare autosomal recessive disorder characterized by defects in LROs, resulting from mutations in one of at least 11 identified genes (HPS-1 to HPS-11).3,4 These gene products assemble into four protein complexes—AP-3 and BLOC-1, -2, and -3—each critical for endolysosomal trafficking and LRO biogenesis. 5 Clinical manifestations vary by genotype but tend to cluster according to shared complex involvement; for example, mutations in HPS1 or HPS4 (components of the BLOC-3 complex) often present with oculocutaneous albinism, bleeding diathesis, and pulmonary fibrosis. Molecular diagnosis of the causative gene is vital for accurate subtype classification, prognostic guidance, and development of targeted therapies.
The reporting of this study conforms to the CARE (Consensus-based Clinical Case Reporting Guideline) guidelines. 6 The CARE checklist is available in the Supplemental Material. Here, we report an unusual case of a 38-year-old women who suffered from gradually aggravated cough and dyspnea in the past 1 year, along with childhood-onset photophobia, photosensitivity, low vision, and repeatedly ecchymosis, who was diagnosed of HPS by platelet transmission electron microscopy. Finally, we identified a novel homozygous mutation of HPS, expanding the molecular epidemiology and genetic counseling databases for HPS. Our case also provides more information of the effectiveness and safety of antifibrotic drug nintedanib in HPS.
Case presentation
Clinical findings
A 38-year-old Chinese women was admitted to our hospital with a 1-year history of dry cough and progressive dyspnea in June 2019. She denied experiencing fever, sweating, weight loss, diarrhea, abdominal pain, dry eyes, dry mouth, joint pain, muscle pain, abdominal discomfort, constipation, gingival bleeding, menorrhagia, et al. She was the only child of nonconsanguineous parents. Her mother died of colon cancer at 72-year-old and her father died in a car accident at 42-year-old. There was no family history of similar symptoms, diseases, or other genetic disorders. She was a nonsmoker and denies occupational dust and organic particles exposure.
On physical examination, the patient presented with hypopigmented hair and fair skin, accompanied by scattered ecchymoses (Figure 1). Auscultation of the lungs revealed fine, velcro-like inspiratory crackles at both lung bases. There was no evidence of central cyanosis at rest while breathing ambient air. Digital clubbing of the fingers and toes was absent. The remainder of the physical examination was within normal limits.

Image of scattered in lower extremities of the patient.
Her serum platelet counts, activated partial thromboplastin time, prothrombin time, international normalized ratio, and fibrinogen values were all in normal range, which indicated normal coagulation function. The screening test for antinuclear antibodies (ANA), rheumatoid factor, antineutrophil cytoplasmic antibodies and anticyclic citrullinated peptide (anti-CCP) antibody, myositis-specific, and myositis-associated antibodies were all negative. The sputum culture for bacteria, fungi, and mycobacteria were negative. Other tests including basal basic chemistry panel and HIV serology test were normal. The high-resolution computed tomography (HRCT) images demonstrated subpleural predominant ground-glass opacities and reticular opacities, along with mild traction bronchiectasis/bronchiolectasis, a radiological pattern of nonspecific interstitial pneumonia (NSIP), seen in Figure 2(a)–(d). Lung function tests revealed mild restrictive ventilatory dysfunction and mildly decreased diffusion function, with a forced vital capacity (FVC) of 2.46 L (76.7% of predicted), forced expiratory volume in 1s (FEV1) of 2.44 L (88.3%), FEV1/FVC 99.30%, total lung capacity (TLC) 4.22 L (86.1%), and diffusion capacity for carbon monoxide (DLCO) 76.7% of predicted.
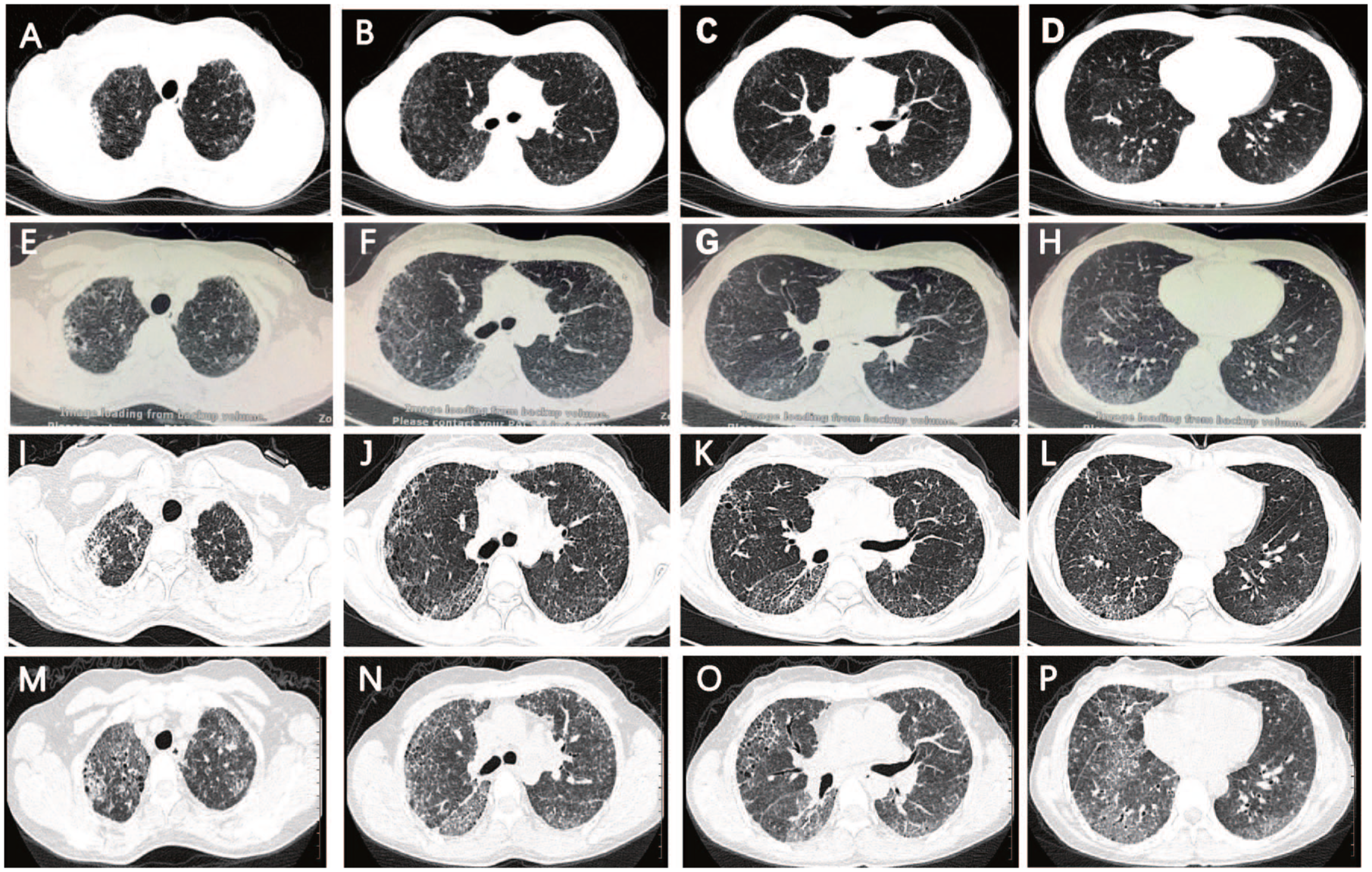
Changes in HRCT findings of our HPS case. (a–d) images obtained at the initial visit, (e–h) images obtained after 12 months of treatment with pirfenidone, (i–l) images obtained after 6 months of treatment with nintedanib switching from pirfenidone, (m–p) images obtained after diagnosis of COVID-19 infection.
As is shown in Figure 3(a) and (b), optical coherence tomography (OCT) imaging revealed foveal hypoplasia, and direct ophthalmoscope demonstrated retinal hypopigmentation.

Ophthalmic examination results. (a) Optical coherence tomography (OCT) scans showed foveal hypoplasia. (b) Fundus photography showed hypopigmented retina.
Diagnostic test
Written informed consent was obtained from the patient prior to participation. Peripheral blood samples were collected for platelet electron microscopy and genomic DNA extraction. The study was approved by the Institutional Review Board of Chengdu Integrated TCM and Western Medical Hospital, in accordance with the Declaration of Helsinki.
Platelet transmission electron microscopy showed almost complete absence of dense-granules, confirming the diagnosis of HPS (Figure 4).

(a) Platelet transmission electron microscopy of the patient showed almost complete absence of dense-granules. Magnification = 15,000. (b) Platelet transmission electron microscopy of health control showed dense-granules (white arrow). Magnification = 15,000.
Genomic DNA analysis by panel sequencing of 31 albinism related genes (AP3B1, BLOC1S3, BLOC1S6, DTNBP1, GPR143, HPS1, HPS3, HPS4, HPS5, HPS6, LYST, MLPH, MYO5A, OCA2, RAB27A, SLC45A2, TYR, TYRP1, MYO7A, KIT, MITF, USH1C, PCDH15, USH1G, CDH23, USH2A, MC1R, GPR98, C10orf’11, EDNRB, SLC24A5) were conducted and the sequencing range covered the exon region and the intron boundaries. Results showed homozygosity for novel deletion mutation in the HPS4 gene (NM_022081.6 c.1713+1delG) on chromosome 22q12.1, and verified by Sanger sequencing, shown in Figure 5(a).
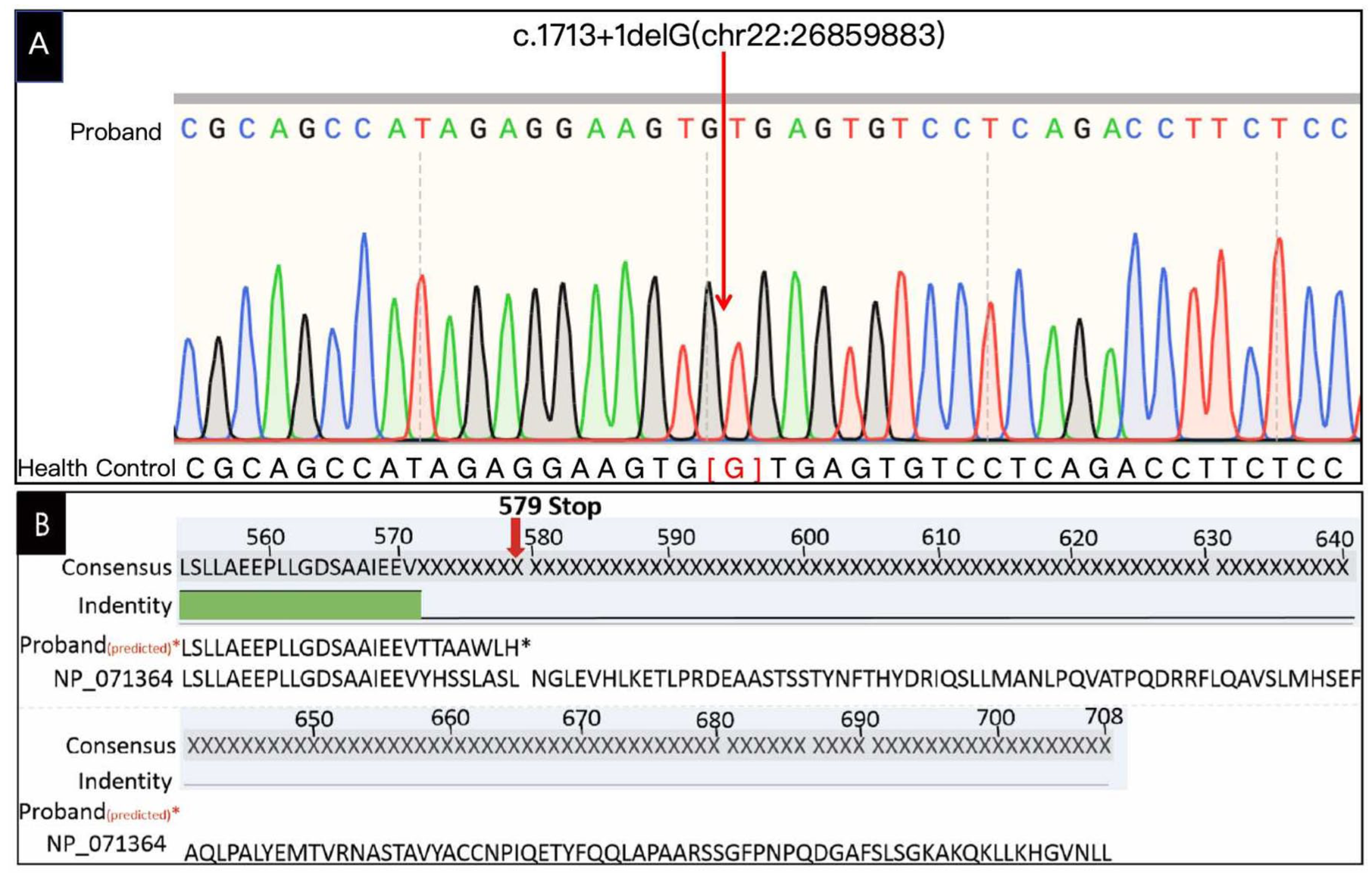
The HPS4 gene mutation identified in the proband and its predicted consequence on the protein sequence. (a) The Sanger DNA sequence chromatogram identifies a novel deletion mutation at the first nucleotide of Intron 11 in the HPS4 gene (NM_022081.6 c.1713+1delG) on chromosome 22q12.1. (b) This splice site variant change causes a frameshift of the amino acid sequence and lead to an abnormally truncated protein.
This variant (c.1713+1delG) was not reported in any of the public databases including ESP6500siv2_ALL, 1000g2015aug_ALL, ExAC, gnomAD and dbSNP147 and was then considered to be novel. To predict the likelihood of pathogenicity, Mutation Taster (http://www.mutationtaster.org) and was employed, with the interpretation of variant guided by the American College of Medical Genetics and Genomics guidelines. 7 The variant was predicted to be “disease-causing” because of splice site change and lead to shorten of amino acid sequence from 708 to 579, shown in Figure 5(b). This splice site change was also predicted by spliceAI with a strong donor loss (DL = 0.99, +2 bp) and a donor gain (DG = 0.96, +3 bp). The mRNA carrying premature termination codons might also trigger nonsense-mediated decay. When using predictprotein (https://predictprotein.org/#) to predict the protein secondary structure, the new protein has less alpha-helix, beta-sheet, and random coils, which may give rise to its dysfunction.
Treatment course
The patient was prescribed with pirfenidone (800 mg, t.i.d.) immediately after diagnosis. One year after starting pirfenidone, her HRCT showed a slowly progressive fibrosis, manifesting as increasing extent and severity of traction bronchiectasis, bronchiectasis, ground-glass opacities, and new honeycombing, as seen in Figure 2(e)–(h). She also suffered from more severe breathing difficulties (FVC: 74.6% predicted, DLCO: 66.2% predicted). After carefully counseling about the potential risks and benefits of nintedanib in treating HPS, the patient determined to change antifibrotic therapy from pirfenidone to nintedanib (150 mg, b.i.d.), while closely monitoring for side effects. After another half a year, she came back for a check with nearly steady HRCT image (Figure 2(i)–(l)) and respiratory symptoms (FVC: 75.4% predicted, DLCO: 70.6% predicted), which led to continuing use of nintedanib for another 1 year. During the whole process of antifibrotic therapy, she had good drug tolerability without obvious adverse drug reactions including bleeding. However, after taking nintedanib for a total of 1.5 years, the patient died of COVID-19–triggered severe respiratory failure, seen in Figure 2(m)–(p).
Discussion
Among 11 subtypes (HPS-1 to HPS-11), HPS-1 disease (MIM 604982) was most frequently reported, with a 16-bp frameshift duplication in exon 15 of the HPS1 gene (604982.0001) to be especially highly prevalent in northwestern Puerto Rico, as the result of an apparent founder effect.8,9 However, the other subtypes of HPS are extremely rare. We report here a Chinese HPS case with a novel homozygous mutation (NM_022081.6 c.1713+1delG) in HPS4, which serves as a valuable addition to the few HPS variants reported to date.
In 2002, Suzuki et al. first identified the human le homolog, HPS4, and reported seven non-Puerto Rican HPS patients with five different variants in this gene. 9 We reviewed published literatures regarding HPS-4 diseases and found a total of 51 cases with 37 variants, including 16 nonsenses, 8 frameshifts, 7 splice substitutions, 5 missenses, and 1 gross insertion, which is shown in Supplemental Table 1.5,10–30 The 37 variants located in Exons 3, 4,5, 6, 7, 8, 10, 11, 13, 14, and Introns 4, 7, 9, 11, respectively. The 51 cases includes 24 Asians (7 Pakistani, 6 Japanese, 5 Chinese, 5 Indian, 1 Sri Lankan), 23 Europeans (3 Turkish, 2 Italian, 2 Ashkenazi-Jewish, 2 Spanish, 1 English, 1 Croatian, 1 Dutch, 1 German, 1 Hungarian, 9 others), 1 Americans (1 Uruguayan), and 3 with unknown ancestry. The high prevalence of this autosomal recessive disorder in Pakistan may be attributed to the high rate of consanguineous marriages, with approximately 60% of the population engaging in cousin unions. 26
In addition to the typical clinical manifestations such as OCA and bleeding tendency, which are common across all HPS subtypes, the phenotypes associated with HPS-4 and HPS-1 are generally more severe due to the frequent complications of colitis and progressive pulmonary fibrosis. 27 According to published reports, the age of patients diagnosed with HPS-4 ranges from 3 months to 61 years. Among those with pulmonary fibrosis, the reported age at diagnosis spans from 19 to 61 years, with associated genetic variants including nonsense, frameshift, splice-site, and missense mutations. In contrast, patients with granulomatous colitis were aged between 13 and 50 years, and harbored primarily missense, nonsense, and frameshift mutations. Notably, the earliest reported onset of pulmonary fibrosis and colitis in HPS-4 patients was at 19 and 13 years of age, respectively—significantly earlier than the typically reported onset in the third or fourth decade of life. 31 Therefore, it is important to monitor for pulmonary fibrosis and colitis in these patients even from their teenage.
The HPS4 mRNA transcript (NM_022081.6) comprises 14 exons and encodes a 708-amino acid protein (~76.9 kDa), known as BLOC3S2 (OMIM: 606682). The functional characterization of HPS4 was first described by Martina et al., who demonstrated that HPS1 and HPS4 form the biogenesis of lysosome-related organelles complex-3 (BLOC-3). Through co-immunoprecipitation of both epitope-tagged and endogenous proteins, they confirmed in vivo assembly of the HPS1–HPS4 complex, which was further shown to be a moderately asymmetric ~175 kDa structure via size-exclusion chromatography and sedimentation velocity analysis. 32 Andreas et al. demonstrated the molecular functions of Hps1–Hps4 complex BLOC-3 to act as the specific Rab32/38 guanine nucleotide exchange factor, which is pivotal for the biogenesis of melanosomes, platelet-dense granules, lamellar bodies, and lysosomes. 33 As a result, the dysfunction of BLOC-3 lead to those cytoplasmic organelles defects, manifesting as OCA, bleeding, and accumulation of ceroid-like materials (pulmonary fibrosis and colitis).
The novel variant (NM_022081.6 c.1713+1delG) is located at the first nucleotide of Intron 11, strongly indicating splice site change and leading to a frameshift with a premature termination codon lead to an abnormally truncated HSP4 protein. Pathogenic variants located within intronic regions should not be overlooked, even though they are often excluded from routine clinical sequencing. To date, seven splice-site variants have been reported in HPS4, four of which occur at the first nucleotide of introns, underscoring the functional relevance of these noncoding regions. Okamura et al. reported an intronic variant (c.596+1G>A ;IVS7+1G>A) on HPS-4 to be detrimental and confirmed the splice pattern change leading to skipping of exon 7, by RT-PCR analysis. 19 Although no additional experimental evidence for function identification is available for other intronic variants in HPS4, the pathogenic mechanism of variants could also be determined by online prediction tools. Jones et al. identified c.597-2A>T of HPS4 in the intron 7 splice acceptor site. In silico prediction suggests this variant cause abnormal splicing between, resulting in a frameshift, a premature stop codon, and nonsense-mediated decay of the variant mRNA. 21 We acknowledge the limitation posed by the unavailability of additional blood samples from the deceased proband and her parents, which precluded functional validation of the genetic variant and prevented construction of a comprehensive pedigree. Nonetheless, the pathogenicity of the identified variant is supported by consistent predictions across multiple in silico tools, thereby reinforcing the reliability of our findings.
Evidence on long-term antifibrotic therapy in HPS remains limited. To date, only two small randomized controlled trials have evaluated pirfenidone. Gahl et al. reported pirfenidone may be effective in progressive pulmonary fibrosis (PPF) due to HPS when forced vital capacity (FVC) is > 50%, whereas Kevin et al. found no statistical difference compared with placebo, leading to early trial termination.34,35 An open-label clinical trial later reported decreased annual decline in FVC and DLCO in HPS patients treated with pirfenidone, with a mean treatment duration of 13.1 years. 36 As a result, pirfenidone is often prescribed on a compassionate basis. While nintedanib is approved for PPF, it is typically avoided in HPS due to bleeding concerns. However, Itano et al. reported the only published HPS-4 case treated with nintedanib, demonstrating approximately 6 months of slowed disease progression without bleeding events. 37 Our case showed similar benefit, with nearly 1.5 years of disease stabilization before the patient succumbed to COVID-19. These cases suggest that nintedanib may be both safe and effective in selected HPS patients. Although lung transplantation has been successfully performed in HPS-related pulmonary fibrosis, it remains inaccessible to many due to high costs and resource limitations. 38 Further research is urgently needed to define optimal treatment strategies in HPS-related lung disease.
Conclusion
In summary, we report a novel homozygous mutation (NM_022081.6 c.1713+1delG) in a Chinese patient with HPS-4, contributing to the expanding molecular and clinical spectrum of the disease and enriching the genetic counseling database. Our findings also provide further support for the potential efficacy and safety of antifibrotic therapy in managing HPS-associated pulmonary fibrosis.
Supplemental Material
sj-docx-1-tar-10.1177_17534666251383677 – Supplemental material for A novel homozygous HPS4 mutation in Hermansky–Pudlak syndrome: case report and literature review
Supplemental material, sj-docx-1-tar-10.1177_17534666251383677 for A novel homozygous HPS4 mutation in Hermansky–Pudlak syndrome: case report and literature review by Qianqian Liu, Wenqi Qing, Shanshan Guo, Yongsheng Wang, Yunfeng Chen and Jie Liu in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-pdf-2-tar-10.1177_17534666251383677 – Supplemental material for A novel homozygous HPS4 mutation in Hermansky–Pudlak syndrome: case report and literature review
Supplemental material, sj-pdf-2-tar-10.1177_17534666251383677 for A novel homozygous HPS4 mutation in Hermansky–Pudlak syndrome: case report and literature review by Qianqian Liu, Wenqi Qing, Shanshan Guo, Yongsheng Wang, Yunfeng Chen and Jie Liu in Therapeutic Advances in Respiratory Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.