
Abstract
Cystic fibrosis (CF) is an autosomal recessive disease caused by the inheritance of two mutant cystic fibrosis transmembrane conductance regulator (CFTR) alleles, one from each parent. Autosomal recessive disorders are rarely associated with germline mutations or mosaicism. Here, we propose a case of paternal germline mutation causing CF. The subject also had an identifiable maternal mutant allele. We identified the compound heterozygous variants in the proband through Sanger sequencing, and in silico studies predicted functional effects on the protein. Also, short tandem repeat markers revealed the de novo nature of the mutation. The maternal mutation in the CFTR gene was c.1000C > T. The de novo mutation was c.178G > A, p.Glu60Lys. This mutation is located in the lasso motif of the CFTR protein and, according to in silico structural analysis, disrupts the interaction of the lasso motif and R-domain, thus influencing protein function. This first reported case of de novo mutation in Asia has notable implications for molecular diagnostics, genetic counseling, and understanding the genetic etiology of recessive disorders in the Iranian population.
Plain language summary
A child can develop Cystic Fibrosis (CF) if both parents pass on mutated genes. In some rare cases, new genetic mutations occur spontaneously, causing CF. This report discusses a unique case where a child has one gene with a spontaneous mutation and inherits another gene mutation from the mother. We used a method called Sanger sequencing to find the two different gene changes in the affected person. We also used computer analysis to predict how these changes might affect the protein responsible for this genetic disease. To confirm that the child's new change is not inherited, we used a type of genetic marker called microsatellite markers. The mutation inherited from the mother and the new spontaneous mutation resulted in a unique change in the responsible protein. This mutation is located in a specific part of the protein called the lasso motif. Our computer simulations show that this mutation disrupts the interaction between the lasso motif and another part of the protein called the R-domain, which ultimately affects the protein's function. This case is significant because it is the first reported instance of a de novo mutation causing CF in Asia. It has important implications for genetic testing, counseling, and understanding how recessive genetic disorders like CF occur within the Iranian population.
Keywords
Introduction
Cystic fibrosis transmembrane conductance regulator (CFTR) is a unique ATP-binding cassette transporter that functions as an ion channel. CFTR consists of 5 domains and 1480 amino acids. 1 Mutations in the CFTR gene are associated with severe pathophysiological conditions in the lungs and pancreatic ducts, leading to cystic fibrosis (CF). 2 This is one of the most common hereditary diseases among Caucasian populations. Therefore, a precise genetic diagnosis is crucial.3,4
Over 2000 CFTR genetic variants are known, 5 with about 350 causing CF. 6 The ΔF508 mutation is the most common, while other pathogenic variants are typically infrequent. 7 Inherited CFTR mutations have been the primary focus of genetic studies in recent decades. De novo mutations in CFTR are sporadic, like other autosomal recessive disorders, and have received limited attention. 8 Germline de novo mutations occur approximately 1.0–1.5 × 10−8 per nucleotide and per generation. 9 A balanced interplay between premutagenic mutations and DNA repair mechanisms results in this low frequency. 10 As a result, only a few reports of de novo mutations in the CFTR gene have been published, with only 10 identified so far.11–17
De novo mutations can arise in parental germline cells or during the early stages of embryonic development in the offspring. 18 Recently, the fitness of mutated premeiotic testis cells, referred to as “selfish spermatogonial selection,” has garnered attention.19–21 As a result, there is growing interest in assessing the likelihood of de novo mutations in spermatogonia using sperm sample analysis. However, this is only feasible in cases where such a sample is available. Haplotype analysis offers the unique opportunity to verify the presence and identify the origin of de novo mutations without the need for sperm samples.22,23
Here, we investigated a family in which the child carried a CF mutation not present in the father (c.178G > A, p.Glu60Lys). Our evidence suggests a paternal germline mutation through the application of phenotypic observations, genetic analysis, and homozygosity mapping of the patient.
Case
Clinical presentation
The proband is a 3-year-old male diagnosed with CF at 2.5 months [Figure 1(a)]. He demonstrated typical CF symptoms such as recurrent coughing and greasy stools with a positive sweat test (first test: Na: 78, Cl: 80; second test: Na: 68, Cl: 70). He also experienced respiratory complications and multiple hospitalizations before the age of 5 months. The developmental patterns were within the expected range, and the patient was regularly examined every 3 months. The high-resolution computed tomography scan of the chest revealed diffuse mosaic attenuation, mild bronchiectasis, and diffuse peribronchial thickening. A potential exacerbation of CF was observed through bronchioles, mucous plugs, and atelectasis in the right upper lobe and inferior lingular segment. The presence of prominent bilateral pulmonary arteries raises the possibility of pulmonary hypertension.

Pedigree and genetic analysis of the studied family. (a) Pedigree and linkage analysis: The pedigree of the family under study is presented, highlighting the relationships between individuals. Sanger sequencing was performed to identify the presence of mutations. Individuals II1 and I2 were found to share the same mutation, as indicated. The linkage analysis also revealed similar haplotypes based on STR markers, represented by corresponding colors. The numbers denote the base pair size of the STR markers, with each marker size ending in the respective digit. The relative location of the c.1408 A > G SNP is shown. (b) The results of Sanger sequencing for the father, mother, and proband were analyzed using the Chromas program. On the left, the Sanger sequencing result of the proband and his father were compared, and it was observed that his father does not carry the c.178 G > A mutation. On the right, the Sanger sequencing result of the proband and his mother were compared, and it was found that both carry the c.1000 C > T mutation.
Since the CF diagnosis, he has regularly consumed pancrelipase delayed-release capsules (250–500 daily units depending on the gastrointestinal health, CREON, Abbott GmbH, Hanover, Germany). Omeprazole (20 mg/day) and Salbutamol (2 × 100 µg/day, air puffs) were prescribed to prevent gastrointestinal and respiratory complications. Additionally, Vitamin E was used as an antioxidant to prevent fatty liver, and daily respiratory physiotherapy and nebulizer treatment (7% NaCl solution w/v) were administered.
The proband’s father and mother were 39 and 34 years old at the time of examination. The mother was 7-month pregnant with the next child at the referral time to our center for a prenatal diagnosis. Both parents are Iranians, unrelated, and they have no previous genetic tests. In this study, we followed the CARE reporting guidelines to ensure accurate and transparent reporting. 24
Genotyping
Genomic DNA was extracted from blood sample and chorionic villus sampling using the salting-out method. 25 In the proband and all family members, the CFTR gene was amplified. Primers were designed to amplify all CFTR gene exons and 200 bp flanking regions. The amplicons were purified using the KBC DNA Pure DNA extraction kit (Kawsar Biotech Co., Tehran, Iran). The BigDye Terminator Cycle sequencing kit (Thermo Fisher Scientific, Foster City, CA, USA, TF) was employed for amplicon sequencing, and a 3130/XL Genetic Analyzer (TF) was utilized for analysis. Bioinformatic tools, including Mutation Taster, 26 Polyphen-2, 27 CADD, 28 FATHMM, 29 SIFT, 30 and PROVEAN, 31 were employed to investigate variant pathogenicity. The 3D structure of the protein was analyzed by modeling in the UCSF ChimeraX software package (version 1.3). 32 Genomic DNA sequencing revealed two mutations in the CFTR gene of the proband that result in a genotype of c.178 G > A/c.1000 C > T. The mother (family member I-2) was heterozygous for the c.1000C > T mutation, while the 7-month-old fetus had two normal CFTR alleles, as depicted in Figure 1(a). Surprisingly, the paternal genotype (family member I-1) appeared normal based on the same sequencing data [Figure 1(b)]. To ensure that the polymerase chain reaction (PCR) conditions were the cause of a missed mutation, we attempted multiple conditions for family member I-1 [Figure 1(a)], such as altering the annealing temperature (Table 1) and location of the sequencing primers. However, the paternal genotype remains normal. Based on the sequencing data, family members I-1, II-1, and II-2 were also heterozygous for the c.1408 A > G polymorphism.
Different PCR conditions and primers for exon 3 of CFTR gene.

CFTR, cystic fibrosis transmembrane conductance regulator.
Haplotype analysis and paternity testing
Paternity testing was performed using the IR-filer PCR amplification kit following the manufacturer’s protocol (Kawsar Biotech Co., Tehran, Iran). Haplotype analysis was conducted using the GT Hapscreen CFTR kit (GENETEK Biopharma GmbH, Berlin, Germany). The fragment analysis was also performed on an ABI 3130/XL Genetic Analyzer (ThermoFisher Scientific, Foster City, CA, United States), and the haplotypes were visualized. We also confirmed the paternity by evaluating 17 distinctive DNA markers (Supplemental File 1), including linked short tandem repeat (STR) markers to the CFTR gene.
Given the confirmed paternity and the lack of c. 178 G > A mutation in either parent, we suggest that this is de novo mutation in the affected child.
Discussion
This study describes the result of a comprehensive genetic analysis in a family with cystic fibrosis (CF). We demonstrated that the affected child carried two mutations in the CFTR gene, with one inherited from the mother. The second mutant allele, c.178 G > A, could not be identified in the father’s genome. Our findings indicated that the c.178 Gs > A, p. Glu60Lys mutation is generated de novo. We propose below that the allele variant stems most likely from a mutation in the spermatogonial cells as compared to germline mosaicism. Notably, this study represents the first report of a de novo mutation occurring in the N-terminal lasso motif of the CFTR protein. To our knowledge, this is the first reported de novo CFTR mutation in Asia.
The affected child presented positive sweat test results and manifested typical CF symptoms, an autosomal recessive genetic disorder. He had the c.1000 C > T, p. Arg334Trp, mutation from his maternally inherited chromosome, as indicated by its presence in the mother’s genome. Gasperini et al. 33 reported c.1000 C > T, R334W mutation for the first time (in a Spanish patient with lung and pancreatic involvement), and it has been reported as a common variant in Iran. 34 Also, a phenotype/genotype investigation of 16 CF patients with R334W demonstrated that symptoms caused by R334W tend to be less severe than those caused by F508 deletion. 35
In addition to R334W, our subject had a missense de novo mutation, c.178 G > A. Our first aim was to understand its impact on the structure of the protein. Currently, there is no report on the clinical symptoms of patients with the p. Glu60Lys mutation in the literature where a glutamic acid is altered in the CFTR protein’s N-terminal of the lasso motif (Figure 2). However, based on cftr2 36 and the SickKids 37 database, p. Glu60Lys leads to impaired lung function and has been reported in both pancreatic-sufficient and pancreatic-insufficient patients. These phenotypes are consistent with the symptoms of our subject.
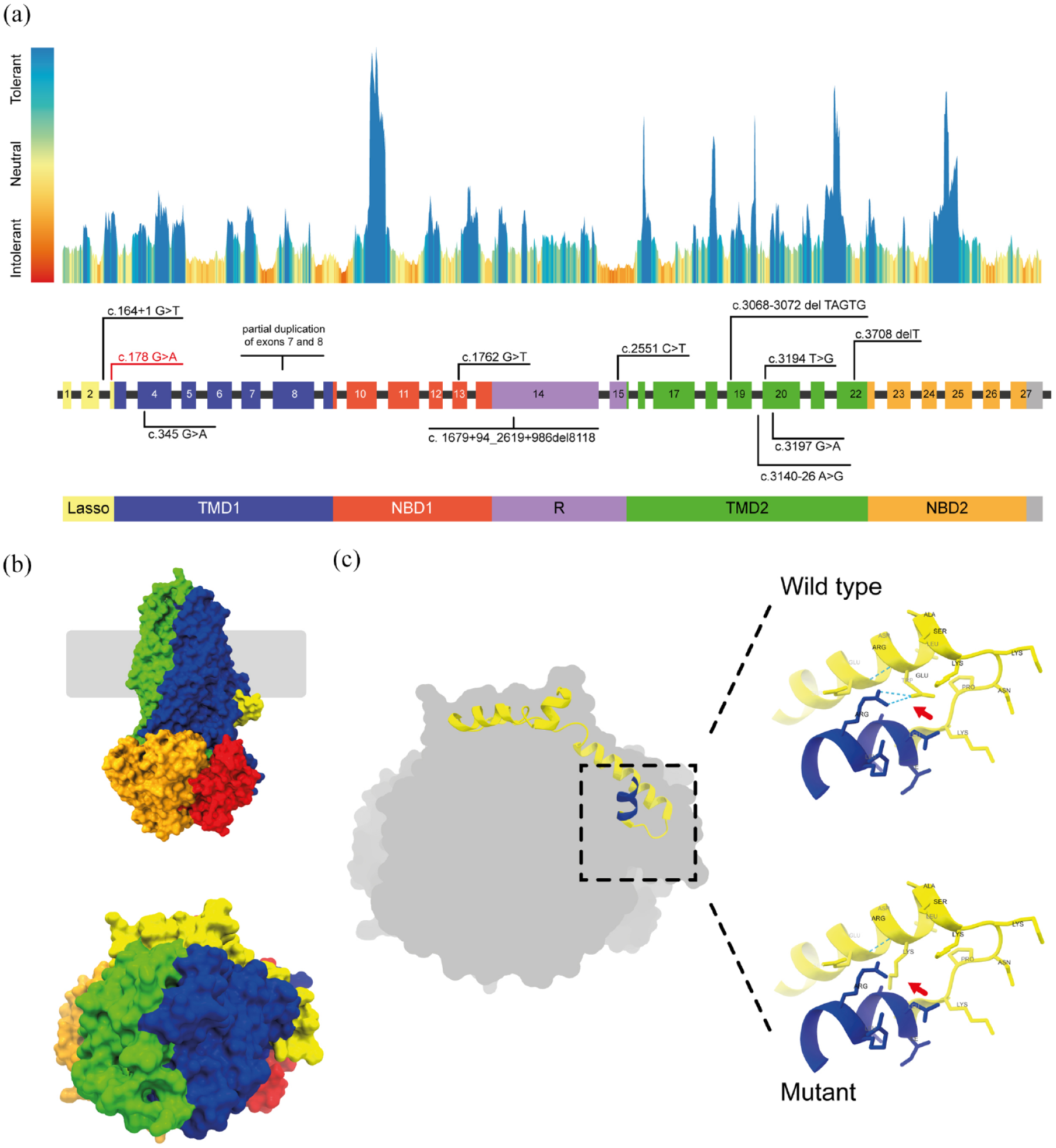
In silico structural analysis of the de novo variant. (a) CFTR exons and protein landscape: The drawing depicts the CFTR exons and protein structure, highlighting the regional tolerance or intolerance to missense or synonymous variation as determined by MetaDome analysis. Known de novo CFTR variants are indicated, while the position of the c.178G > A mutation is highlighted in red. Different colors represent specific protein domains with corresponding exon colors. Gray areas represent protein regions where the domain border is undefined. (b) Structural model of human CFTR protein: A structural model of the human CFTR protein (PDB: 6MSM) is presented, providing a visual representation of its three-dimensional structure. (c) Predicted effects of E60K mutation: On the right and top, the Glutamic acid residue (Glu60) is shown, indicating the presence of a hydrogen bond (dashed lines) between Glu60 and Arg75. The substituted Lysine residue (Lys60) is demonstrated on the right and bottom, illustrating the absence of a hydrogen bond between Lys60 and Arg75. These visualizations show the predicted impact of the E60K mutation.
Several studies have reported patients with the homozygote R334W genotype,38–40 while the E60K mutation has only been reported in compound heterozygote forms.41–43 To investigate the effects of E60K mutation on the CFTR protein, Gene et al. expressed this variant in the HEK 293 cell line and conducted functional studies. They reported that the E60K mutation results in a misfolded CFTR protein that undergoes degradation in the cytoplasm, similar to the more common F508del deletion variant. 44 Glutamic acid at position 60 is the endpoint of a highly conserved amino acid sequence involved in PKA-dependent channel gating through interaction with the R-domain.45,46 The substitution of glutamic acid for lysine likely disrupts this interaction and impairs the protein function. Substitution of a negative charge (glutamic acid) for the positive (lysine) might also cause repulsion among the protein core residues.
Additionally, glutamic acid at position 60 forms a hydrogen bond with arginine at position 75. The larger size of the mutant lysine may impact the positioning of this residue, which would need a new hydrogen bond partner [Figure 2(b)]. The larger mutant residue also likely affects the structure by disrupting the proper folding of the protein core.
The affected child in our study was the only carrier of this mutation in the whole family. Interestingly, his father’s genome did not carry these mutations. We investigated three possibilities of nonpaternity: allele dropout during PCR, or a de novo mutation. First, the paternal relationship was confirmed through haplotype analysis, confirming family member 1-I was, in fact, the biological father (data not shown). To rule out allelic dropout, we modified PCR conditions and designed new primers. Sequencing the amplified DNA from the father yielded no mutations in the CFTR gene, eliminating allelic dropout as a potential explanation. Given the three possible hypotheses, we conclude that the mutation is de novo.
Geneticists have long been interested in investigating the features and prevalence of de novo mutations. These mutations are typically inherited from the paternal lineage with increased likelihood in older fathers.47,48 Some genomic regions are more susceptible to mutation, and several factors influence mutation rates, such as replication timing and local base pair combinations. 49 The c.178 G > A is located in the third exon of the CFTR gene; also, another nonsense mutation, c.178 G > T, is reported in the same position. 48 The region spanning cDNA nucleotides 166–182 harbors ten mutations. The high number of mutations in this region and an increased likelihood of a de novo C: G > T: A transition in non-CpG sites might explain the nucleotide alteration at this position. 50
We found the c.1408 heterozygous polymorphism in the affected child, father, and fetus, which we used as a complementary marker for homozygosity mapping and mosaicism analysis. STR markers revealed a crossover in the paternally inherited chromosome of the fetus [see Figure 1(A)]. Since the fetus inherited the closely linked c.1408 A > G polymorphism to the c.178 position, our subject likely inherited the same allele from the father, which supports the hypothesis of a de novo germline mutation. Therefore, we can conclude that this is most probably a de novo mutation originating during spermatogenesis and not gonadal mosaicism. Nevertheless, gonadal mosaicism can only be ruled out by either having another child or obtaining gonadal cells like sperm from the father. Unfortunately, neither was possible since the family did not have any other child, and the father did not agree to give sperm due to religious reasons. It is also possible that the de novo mutation appeared during the early stages of embryonic development.
Conclusion
We investigated a family with a cystic fibrosis patient and discovered the first reported de novo mutation in the N-tail lasso motif of CFTR and the first identified de novo CFTR mutation in Asia. This study highlights that STR markers can play an essential role in determining the developmental origin of the mutated chromosome. Moreover, these findings have significant implications for genetic counseling and prenatal diagnosis. It provides evidence that de novo mutations in cystic fibrosis and other autosomal recessive disorders may be more prevalent than expected, as demonstrated in Spinal Muscular Atrophy (SMA) patients previously. 51 Thus, it is possible, through de novo mutations, to have an affected child with only one parent carrying a single recessive allele. However, it is also crucial to rule out confounders such as paternity and PCR allele drop-out and not to assume that every mutation is de novo.
Supplemental Material
sj-docx-1-tar-10.1177_17534666241253990 – Supplemental material for Identification and in silico structural analysis for the first de novo mutation in the cystic fibrosis transmembrane conductance regulator protein in Iran: case report and developmental insight using microsatellite markers
Supplemental material, sj-docx-1-tar-10.1177_17534666241253990 for Identification and in silico structural analysis for the first de novo mutation in the cystic fibrosis transmembrane conductance regulator protein in Iran: case report and developmental insight using microsatellite markers by Amin Hosseini Nami, Mahboubeh Kabiri, Fatemeh Zafarghandi Motlagh, Tina Shirzadeh, Hamideh Bagherian, Razie Zeinali, Ali Karimi and Sirous Zeinali in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-jpg-2-tar-10.1177_17534666241253990 – Supplemental material for Identification and in silico structural analysis for the first de novo mutation in the cystic fibrosis transmembrane conductance regulator protein in Iran: case report and developmental insight using microsatellite markers
Supplemental material, sj-jpg-2-tar-10.1177_17534666241253990 for Identification and in silico structural analysis for the first de novo mutation in the cystic fibrosis transmembrane conductance regulator protein in Iran: case report and developmental insight using microsatellite markers by Amin Hosseini Nami, Mahboubeh Kabiri, Fatemeh Zafarghandi Motlagh, Tina Shirzadeh, Hamideh Bagherian, Razie Zeinali, Ali Karimi and Sirous Zeinali in Therapeutic Advances in Respiratory Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.