
Abstract
Idiopathic pulmonary fibrosis (IPF) is a devastating, progressive and ultimately fatal lung disease. The combination of poor prognosis, uncertainty of disease course and severe symptom burden heavily impacts patients’ and their families’ quality of life. Though new antifibrotic drugs have been shown to decrease disease progression, the effect on health-related quality of life (HRQOL) has not been convincingly demonstrated. In a relentless disease such as IPF, striving to optimize HRQOL should complement the endeavour to prolong life. Unfortunately, there is a paucity of interventions improving symptoms and functionality for patients with IPF, and research focusing on symptom improvement, and assessing and optimizing HRQOL, is limited.
This review summarizes the most recent insights into measuring and improving quality of life for patients with IPF, and discusses challenges in the management of this devastating disease. Moreover, we postulate a new model for continuous care in IPF – ‘the ABCDE of IPF care’:
Introduction
Idiopathic pulmonary fibrosis (IPF) is a devastating, progressive, fibrotic lung disease. IPF is characterized by irreversible loss of lung function, ultimately resulting in most patients dying from respiratory failure within 3–5 years. The most common symptoms are dry cough, dyspnoea and fatigue. 1 Though the disease course may vary among patients and many patients experience periods of relative stability, disease progression and worsening of symptoms are inevitable for the majority of patients.2,3 The combination of poor prognosis, uncertainty of disease course and severe symptom burden heavily impacts quality of life (QOL) both for patients and family members.1,4,5 Recently, knowledge on the pathogenesis of the disease has improved and has led to the development of two antifibrotic drugs that slow down disease progression as measured by decline in pulmonary function. Though these drugs are major steps forward for patients, IPF still remains a devastating and deadly disease. It is encouraging that multiple new compounds are currently investigated in clinical trials, aiming at modifying the disease course. The majority of these trials are focused on disease modification measured by physiological parameters, such as lung function. However, in many patients with IPF, there is no clear correlation between physiological parameters and patient-reported outcome measures (PROMs), such as health-related QOL (HRQOL) scores, that strive to reflect how a patient feels or functions. In a relentless disease as IPF, striving to optimize HRQOL should complement the endeavour to prolong life. There is a paucity of interventions that convincingly improve symptoms and functionality for patients with IPF. This review summarizes the most recent insights into measuring and improving QOL in patients with IPF and postulates an ABCDE of IPF care, to facilitate a systematic and comprehensive approach to care for patients with IPF.
Health-related quality of life in IPF patients
Health-related quality of life refers to a person’s satisfaction with aspects of life that may be affected by health. 6 As clinicians, we focus primarily on HRQOL since QOL is also determined by such aspects of life as freedom, quality of environment and financial situation. Nevertheless, with a relentless disease as IPF, almost all aspects of life can become health-related. 6 Patients with IPF report an impaired HRQOL. 1 Symptoms as dyspnoea, decreased mobility and cough are often important determinants of HRQOL.7,8 HRQOL is generally assessed by patient-reported outcome measures. 9 Often, there is a poor correlation between physiological assessments of disease severity by pulmonary function testing and patient-reported outcomes of HRQOL and symptoms. 10 Drugs that may modify the disease, by slowing down the pace of lung function decline, do not convincingly improve patients’ overall symptoms or HRQOL.11,12 Therefore, complementary disease management strategies are needed to improve HRQOL.
Unmet needs of IPF patients and caregivers
Several recent initiatives have examined the needs of patients with IPF and their relatives.1,5,13–18 Though identifying patient-specific needs is crucial in the individual treatment relationship between a patient and care provider, it should be recognized that many basic conditions for IPF care are still frequently unmet. An initiative by 11 European patient advocacy groups for pulmonary fibrosis identified five key themes of unmet needs: 1) better diagnosis, 2) better access to different treatment forms, 3) availability of emotional support, 4) improved information resources, and 5) equal availability of palliative and end-of-life care. 18 The European IPF Patients Charter was developed based on these outcomes (http://www.ipfcharter.org/call-to-action/).
An interesting study by Overgaard and colleagues not only looked at the unmet needs of patients with IPF, but also included family caregivers. 5 They studied patients’ and caregivers’ experiences of living with IPF, using extensive interviews of patients and their caregivers. In total, 25 patients and 24 family caregivers participated in the study. The main findings of their study showed a need for stepwise information and disclosure, and awareness of differences in reactions and wishes between patients and family caregivers.
A study by Russell and colleagues underscores the previous studies, pointing out a need for better diagnosis of IPF, access to high-quality information on IPF, and emotional support for both patients and family caregivers. 19 Additionally, they found a need for better access to interstitial lung disease (ILD) specialist nurses, and highlighted the meaningful position of physicians and ILD specialist nurses as main contact persons for patients.
Sampson and colleagues looked at the care needs of IPF patients and their carers in different phases of disease course. 17 ‘Carer’ was defined as ‘a person of the patient’s choice who contributed most to their care or, in the early stages of disease, provided emotional support’. Their study shows that although patients and carers had adequate knowledge of the overall prognosis of IPF, it was difficult for them to translate this knowledge regarding their own disease course and the corresponding psychological and physical treatment possibilities. It also recommended that patients and carers needed a different approach to evaluating IPF in clinic, and that physicians should focus not only on lung function parameters, but also on overall health status, self-management, nutrition and an explanation of disease progression.
An underrecognized problem, but with great impact on QOL, is the presence of sexual dysfunction in some patients with IPF. Erectile dysfunction has been associated with chronic obstructive pulmonary disease (COPD) and was reported to be a common problem in ILD.20,21
As a part of the US Food and Drug Administration’s (FDA) patient-focused drug development program, a meeting was conducted with patients to examine their perspectives in treatment approaches. This meeting underscored the need for better medication and symptom relief, in particular for shortness of breath, severe cough and fatigue. 8
Numerous other studies have evaluated the unmet needs of patients with IPF and their family caregivers.13,14,16,22 All confirm the above-mentioned needs, and show the importance of holistic complementary care, focused on optimizing QOL in patients with IPF and their family caregivers.
Patient-reported outcome measures
A variety of tools are used to assess the impact of disease on QOL in IPF; however, there is a paucity of specific well-validated PROMs and a lack of consensus on which tools to use for care and research.23,9 Patient-reported outcomes (PROs) are defined as ‘any report coming directly from the patient without interpretation by a third party about how they feel or function in relation to a health condition and given intervention’. 24 The most commonly used PROMs in IPF are questionnaires originally developed to assess HRQOL in other chronic (respiratory) diseases. These questionnaires have been modified and revalidated for the IPF population. Two of these questionnaires, often used in IPF studies, are the Saint George Respiratory Questionnaire (SGRQ) and the Medical Outcomes Study 36-item short-form health survey (SF-36).25,26 Both are well validated and have proven to possess validity in assessing HRQOL in IPF.27,28 Despite these qualities, they have failed to convincingly show changes in HRQOL in the major IPF trials. The SGRQ has only shown some significant positive changes in the STEP-IPF trial (using sildenafil) and the INPULSIS 2 trial (using nintedanib).
Since a few years, disease-specific PROMs have been developed and validated in IPF patients. A modified IPF version of the SGRQ (SGRQ-I) was created by statistical analysis (Rasch analysis) of the SGRQ data of a clinical trial in IPF. 29 The SGRQ-I holds validity and reliability comparable with the original SGRQ, 29 but the experience with the questionnaire is limited. The King’s Brief Interstitial Lung Disease health status questionnaire (K-BILD) was developed in a population of 124 patients with mixed ILD’s and 49 patients with IPF.10,30 The questionnaire is short (15 questions) and holds good psychometric properties. A Tool to Assess Quality of Life in IPF (ATAQ-IPF) was developed in a group of 95 IPF patients. 31 The questionnaire contains 43 items and is validated in the UK and the US. 32 Longitudinal studies on the performance of the K-BILD and ATAQ-IPF are currently underway.
In addition, regarding IPF several symptom-specific questionnaires have been used. The most commonly used is the University of California San Diego Shortness of Breath Questionnaire (UCSD), which assesses dyspnoea. 33 One of the IPF trials using the USCD was the ASCEND trial (using pirfenidone). Though patients in this trial experienced a significant reduction in their decline of lung function, no effect in UCSD scores was found. 11 The Medical Research Council dyspnoea scale was found to be predictive of disease progression, but its value to assess response to interventions in IPF is unclear. 34 The same limitation applies for other dyspnoea scoring tools, such as the Borg Rating of Perceived Exertion Scale and the Baseline Dyspnoea Index.35,36 Though, by some estimates, over 80% of IPF patient’s experience cough, PROMs on cough in IPF are limited. Both the Leicester Cough Questionnaire (LCQ) and the Cough Quality of Life Questionnaire have been evaluated in IPF, but need further longitudinal validation.37,38 Visual analogue scales (VAS) are also used to assess the severity, frequency and impact of cough. 39 LCQ and cough-VAS show correlation with objective cough counts.40,41 Though fatigue is often a problem in IPF, to our knowledge no validated fatigue PROMs exist for IPF. Anxiety and depression are estimated to be present in around 25% of patients with IPF and are important to recognize.4,42 Although no IPF specific tools exist to screen for anxiety and depression, in general practice common tools such as Hospital Anxiety and Depression Scale and the Centre for Epidemiologic Studies Depression Scale are often used.42,43 It is important to realize that though PROMs were initially mostly developed for use in clinical trials, their use is much broader. Using PROMs in routine care can improve communication, detect unrecognized needs and problems, and serve as outcome measures for interventions.44–46
Optimizing quality of life
Symptoms, perceptions and reactions all interact, and together they influence HRQOL for patients with IPF (Figure 1). This interacting balance wheel of symptoms, perception and reaction varies among patients, but also often changes within individual patients during the disease course (Figure 1).
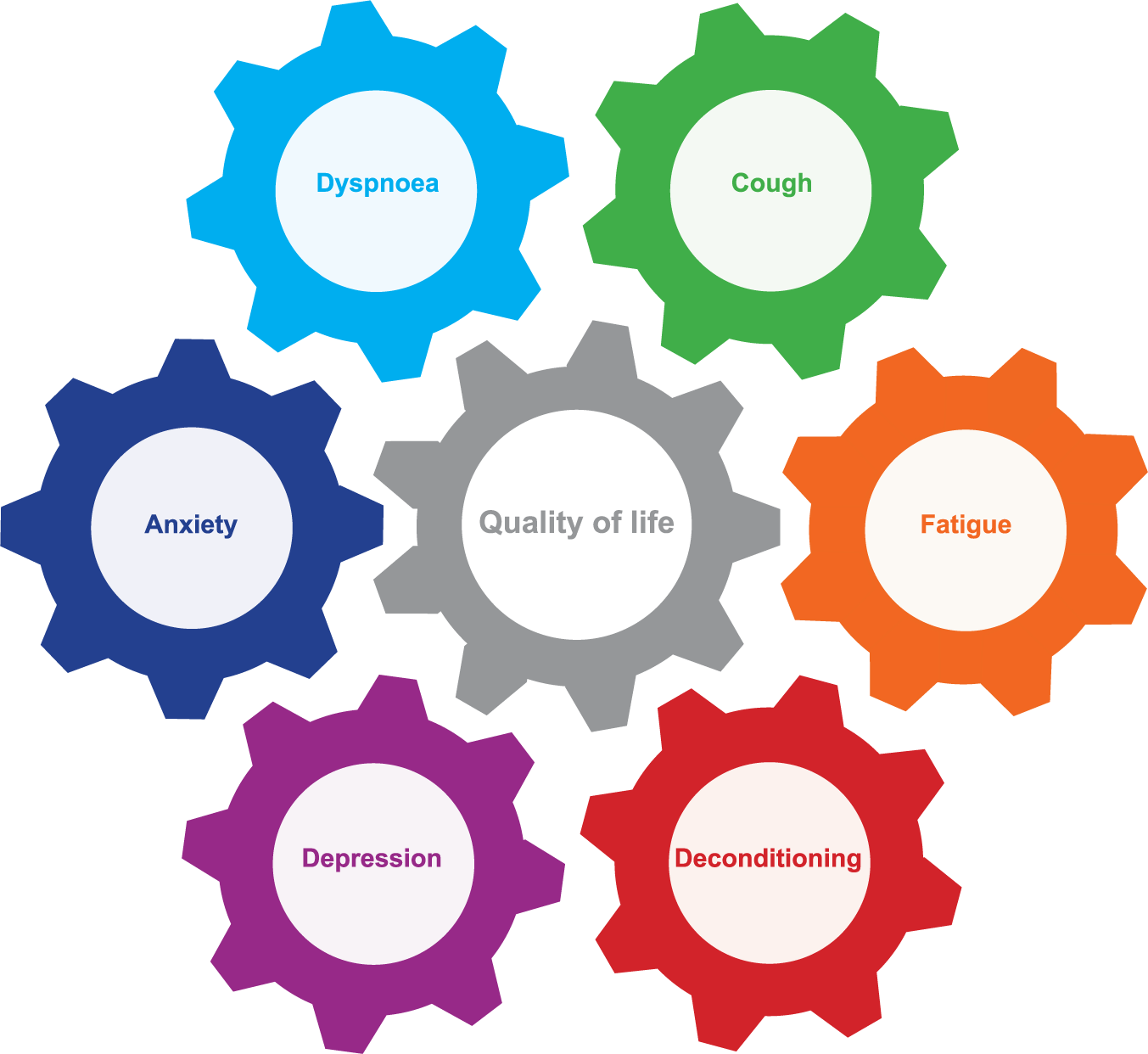
Balance wheel of symptoms, perception and reaction in patients with idiopathic pulmonary fibrosis.
A synchronized comprehensive management strategy is vital to match patients’ needs throughout the disease course. 47 Below, we focus on interventions and treatments that may have a positive effect on HRQOL in IPF. To facilitate a systematic and comprehensive approach to IPF care, we postulate to use an ‘ABCDE of IPF care’ (Figure 2).
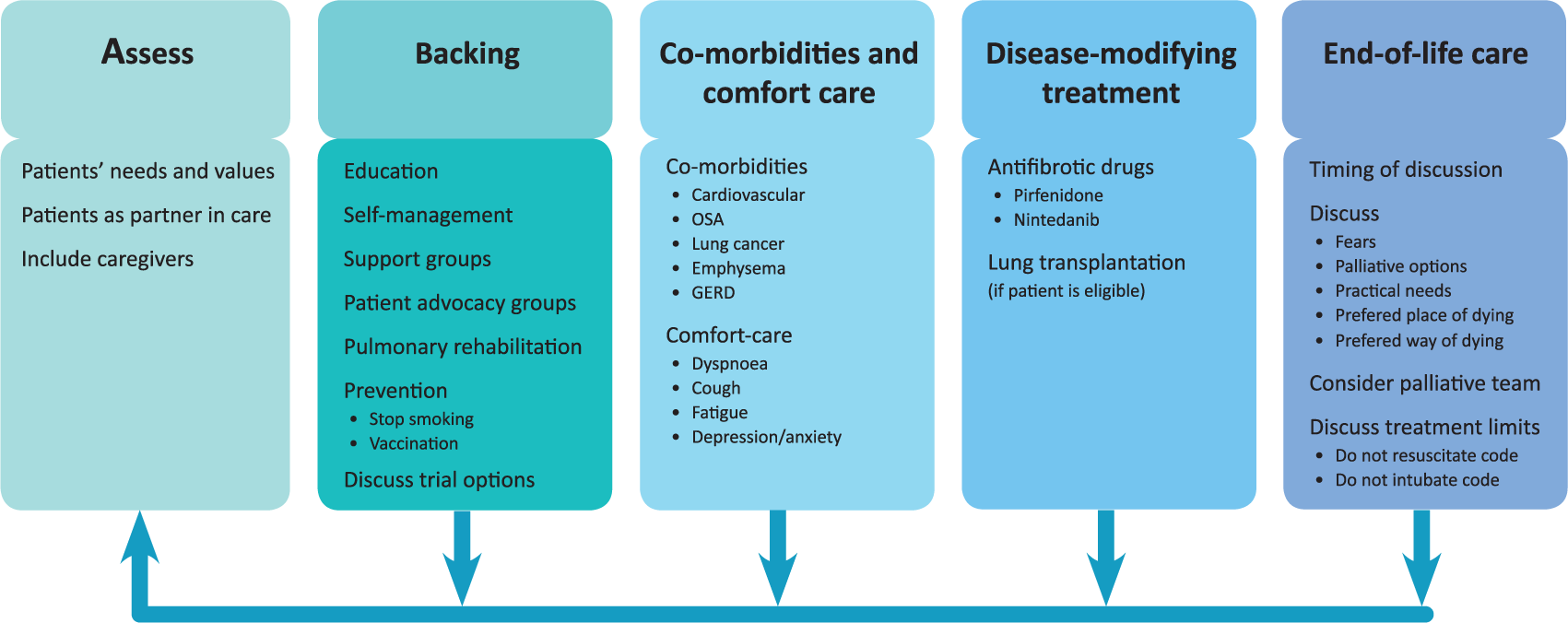
ABCDE of IPF care.
Assess patients’ needs and values
At time of diagnosis, careful discussion of preferences and needs of care should commence, allowing the patient and family caregivers time and space to cope with the diagnosis and information given. Not only will individual patients have different needs and preferences, the success of therapies and interventions will also depend on patient factors such as expectations, experiences and motivations. Continuous reassessment of the individual patient’s wishes and the adaptation of interventions is crucial. Good collaboration between patients and health care providers is the foundation for good care. Both need to trust and respect one another. The patient is obliged to inform the health care provider correctly, while the health carer is committed to provide the best care possible for this patient. Lee and colleagues elegantly modelled this patient–provider relationship as the foundation of care of the IPF patient. 47 Family caregivers should be involved from the onset of the disease and supported as they are the main support for the patient.5,48
Backing patients
Education is one of the crucial factors that empowers patients to make realistic choices and to play an active role in their care. The need for information is universal for patients with IPF and their families, and is iterated in all initiatives on identifying patient needs.5,18,19 Nowadays, many patients diagnosed with IPF and their relatives turn to online sources of health information. Fisher and colleagues showed that these online sources are frequently of poor quality, outdated, or not available in the patients’ native language. 50 In daily practise, clinicians and ILD specialist nurses play a central role in providing information and guiding patients to sources of information and support. Though information is such a crucial factor, research on best practices of educating patients and partners is scarce. Some recent studies underlined that information should be gradually paced, and dyssynchrony between patient and partners in coping with the disease should be taken into account.5,50
Over the last several years, support groups for patients with IPF have expanded (http://www.pulmonaryfibrosis.org/life-with-pf/support-groups). Support groups can decrease anxiety and depression, are helpful in educating patients and family caregivers, and can improve wellbeing. 51 In oncology, support groups have shown their merits in improving QOL.52,53 In IPF, unfortunately, only few studies have looked at the effect of support groups on the QOL of patients and family caregivers. Lindell and colleagues found that a 6-week disease-management program, surprisingly, decreased HRQOL and increased anxiety scores in IPF patients. 54 Nevertheless, stress scores declined in their partners, and all participants found participation beneficial. Contrary to the study of Lindell and colleagues, unpublished data shows that a 3-week multidisciplinary patient and partner empowerment program for IPF (PPEPP) improved QOL. 55 Patient advocacy groups can also play a role in providing information, support and contact with other patients to share experiences. Besides this, patients should be advised on preventative measures. Although no convincing evidence exists on the benefits of vaccination in IPF, influenza and pneumococcal vaccinations are generally advised. If patients still smoke they should be strongly encouraged to quit smoking.
Trials remain crucial to advancing mechanistic insights and stimulating the development of better therapies and intervention aimed at disease modification and improving HRQOL in IPF. A national survey of pulmonary fibrosis patients and family caregivers presented at the PFF Summit 2015 showed that healthcare providers discussed the trials that were currently being conducted with only 55% of their patients (http://www.viddler.com/v/197d1e49). Collaboration between patients, family caregivers and researchers is essential to advance care in IPF. Moreover, information about ongoing research projects and registries should be made available to enable patients to make choices on possible participation in clinical trials and registries.
Comorbidities and Comfort care
Adequate symptom relief is another crucial aspect in optimizing the QOL of patients and family caregivers. 47 IPF is a disease with a high symptom burden for patients. Symptoms often escalate due to the progressive nature of IPF. In patients with IPF, comorbidities are frequent and may importantly contribute to symptom burden and QOL. 56 Identification and treatment of comorbidities may improve QOL and potentially influences prognosis. 57 In the next section, we will review measures that may positively influence the most common symptoms patients with IPF experience; dyspnoea, cough, fatigue/deconditioning and depression/anxiety. 8
Dyspnoea
Dyspnoea is defined by the American Thoracic Society (ATS) as ‘a subjective experience of breathing discomfort that consists of qualitatively distinct sensations that vary in intensity’. 58 Almost all patients with IPF experience progressive dyspnoea, resulting in a major impact on their QOL; living with breathlessness impacts all aspects of daily life. Anxiety and avoidance of exertion lead to deterioration of functional impairment and social limitations. 59 Dyspnoea and change in dyspnoea is also an independent predictor of survival.34,60,61 and has a complex pathophysiology. Possibly, numerous factors – such as mechanics related to the disease as well as psychological and neurological factors – play a role. 58 Hence, it is useful to understand that hypoxemia does not always result in dyspnoea sensations in patients with IPF.
Data on managing IPF-related dyspnoea is scarce. It is important to first rule out comorbidities, such as pulmonary hypertension, cardiac disease, muscle weakness, sleep disorders and psychosocial factors. Although antifibrotic therapies have a disease modifying effect and slow down decline in lung function, they do not reduce dyspnoea. There are a few small scale studies suggesting a beneficial effect of supplemental oxygen on dyspnoea and exercise capacity in patients with IPF.62–64 However, supplemental oxygen can also decrease patients’ QOL as it may restrict daily activities, can be expensive, and makes the disease more visible. 65 Some patients regard the initiation of oxygen therapy as a negative landmark in their disease and, as such, often try to postpone start-up. To increase acceptability, compliance and efficacy of supplemental oxygen, careful attention should be paid to the delivery system (e.g. pulse versus continuous, portable versus home-based).
Pulmonary rehabilitation is widely used to improve exercise capacity in patients with chronic disease. In IPF, pulmonary rehabilitation programmes have also been shown to improve dyspnoea, QOL, physical activity and body composition.66–68 Unfortunately, improvements seem to decrease after finishing the program. Therefore, patients should be encouraged to attend a pulmonary rehabilitation maintenance programme, which may prolong the positive effect of participating.
There is a lack of good quality studies on the effect of opioids on dyspnoea symptoms in patients with IPF. The few existing studies show systemic opioids may have a favourable effect on patients’ experience of dyspnoea. 69 As opioids are often seen as end-of-life care and may also have unfavourable side effects, such as constipation and sleepiness, conscientious explanation of their use and anticipation of side effects is recommended. Colman and colleagues demonstrated in a small study that opioids can also be safely prescribed to those patients with ILD on the waiting list for lung transplants. 70 In a cohort of 38 patients taking chronic opioids for their dyspnoea, there was no respiratory depression and no clinically important opioid toxicity. Furthermore, they observed a trend toward increased exertion during exercise sessions with opioids versus pre-opioids. ATS, for example, recommends opioids as a treatment option for patients with chronic respiratory disease, but also, advises discussing drug choice and dosing with patient and family caregivers beforehand. 71
There are some indications that sildenafil might decrease dyspnoea and improve QOL in patients with IPF. 72 However, debate on this exists and more research is needed. 73 A simple intervention that might be beneficial for patients with IPF is the use of a handheld fan. Booth and colleagues studied the feasibility of this fan in patients with chronic refractory breathlessness and found a decrease of symptoms in half the patients. 74
Cough
No reliable data are available on the prevalence of cough in IPF. Almost 80% of patients with IPF experience some chronic cough, but the number of patients with severe disabling cough may be lower.12,75 Nevertheless, cough is an invalidating symptom that can evoke spells of severe breathlessness and anxiety and may greatly impact patient’s social participation. Cough in IPF has been associated with disease progression. 75 The exact underlying mechanism of cough in IPF patients is unknown, but is most probably ‘multifactorial’ and driven by mechanical, biochemical and neurosensory changes, with an important role for comorbidities as well.76–78
In the treatment of chronic cough in IPF, it is important to first exclude and treat underlying comorbidities. Frequent comorbidities are gastro-esophageal reflux disease, obstructive sleep apnoea (OSA), chronic sinusitis, emphysema, lung cancer, infection and COPD associated chronic bronchitis. Angiotensin-converting-enzyme inhibitors can also cause chronic cough directly after starting the medication, or even months later. 79 IPF-related cough is frequently refractory to antitussive therapy, and the management of cough in IPF often consists of trying different treatment approaches and can thus be frustrating for both patients and clinicians. Low dose steroids are regularly tried for cough in IPF, though there is little data to support their effect. 76 Considering the potential side effects and the fact that steroids as primary treatment for IPF have been associated with worse outcome, risk and benefits should be carefully balanced. In other respiratory diseases, opioids have been shown to decrease cough, although such evidence is absent in IPF data. Thalidomide has been shown to decrease cough in a small 24-week double blind study. 39 As only 20% of screened patients completed the study and thalidomide has a severe side effect profile, we would not recommend it as routine treatment. Recent findings suggest that the antifibrotic drug pirfenidone might have a positive effect on cough. 80 As for nintedanib, its effect on cough is still unknown. While there are no convincing data on over-the-counter cough suppressants in IPF, some patients do report relief from these agents and the risks of trying them are negligible.
Fatigue and deconditioning
Many patients with IPF experience fatigue. The aetiology is multifactorial with factors such as deconditioning, reduction of skeletal muscle strength, cough, dyspnoea and hypoxemia likely to contribute. Additionally, comorbidities and psychological factors may play a role. Fatigue leads to fewer daily physical activities, resulting in a further decrease in exercise tolerance and muscle strength, which, in turn, increases the level of dependency and immobility, negatively affecting HRQOL and social participation. To objectively measure exercise capacity, a 6-minute walk test or cycle ergometry can be done. 81
In the treatment of fatigue and deconditioning, it is again important to rule out comorbidities such as cardiac disease, depression and OSA. Referral to pulmonary rehabilitation is, as for dyspnoea, recommended as treatment for fatigue and deconditioning in IPF patients. Treatment is difficult, as patients find it hard to exercise due to their fatigue and dyspnoea. A decrease in exercise then provokes a downward spiral where muscle strength declines and leads to even less exercise. 82 Early referral is advised, as patients probably benefit most when there they are still able to exercise at full power. 83 In addition, oxygen supplementation can be given when hypoxemia limits exercise capacity; however, the exact benefits of oxygen therapy for fatigue in IPF patients are still unclear. 64 The effects of pharmacological therapy for fatigue symptoms in IPF patients is unknown. 47
Anxiety and depression
In general, patients with chronic diseases are more susceptible to such symptoms as anxiety and depression. Also, many patients with IPF and their partners experience these symptoms.48,84 In ILDs, percentages vary from 7–49% for clinical meaningful depression, and 9–12% for clinical meaningful anxiety.4,43,85,86 There is a known relationship between anxiety and breathlessness in other chronic diseases.87,88 Breathlessness causes anxiety, as patients panic when they cannot breathe and fear the next attack. But anxiety can, on the other hand, increase the perception of breathlessness. 59 It is stressful for family caregivers that they cannot help their loved ones during these attacks. 22 Coughing can increase feelings of anxiety as it induces breathlessness as well. IPF patients also experience anxiety and depression due to the side effects of medication and the uncertainty about the disease course in the context of a poor prognosis. Depression and anxiety are not only essential in predicting the QOL for ILD patients, but can also aggravate breathlessness.42,84 Therefore, screening for depression and other underlying symptoms, that can increase psychological stress and may decrease the patients QOL, is needed. 42
In the treatment of anxiety and depression, it is important to look at comorbidities such as OSA, fatigue and polypharmacy. Again, referral to pulmonary rehabilitation can be beneficial.13,89 A valuable addition to pulmonary rehabilitation might be cognitive behavioural therapy (CBT). 90 CBT focuses on the relationship between thoughts, emotions, physical symptoms and behaviour, and enables people to cope with their negative thoughts. 91 CBT has been shown to decrease anxiety and depression symptoms in patients with COPD and might be beneficial for all chronic physical diseases.42,91,92 In addition, ILD specialist nurses can play an important role in managing such symptoms as depression and anxiety. Since they are readily accessible and closely involved in the patient’s disease path, it is easier for patients to talk to ILD specialists about their concerns and problems than doing so during their short visits to the doctor. Support groups can also help patients with their emotional struggles and are important for educating patients and their family caregivers. 51 The effect of pharmacologic treatment for anxiety and depression, such as antidepressant medication, has not been studied in IPF; thus, the need to use such treatment should be considered thoroughly for each patient.
Disease-modifying treatment
The availability of two antifibrotic drugs with confirmed positive treatment effects has significantly changed the course of the disease and the hopes for patients.11,12 Unfortunately, neither of these drugs cures IPF or completely halts disease progression, and lung transplantation remains the only curative treatment for the small minority of patients who are eligible for this major intervention. Although neither antifibrotic drug has convincingly demonstrated a positive effect on HRQOL, there is no evidence of negative reactions from side effects. It might well be that better tools are needed to detect changes in HRQOL. On the other hand, we should also consider the meaning of nonsignificant changes or stabilization of HRQOL for patients suffering from a disease that remains progressive. Furthermore, it might well be that disease modifying agents do not necessarily improve a patient’s HRQOL. Since HRQOL is determined by many aspects of life and disease, it is evident that complementary treatment strategies are needed.
In the era before the availability of antifibrotic drugs, treatment goals gradually shifted during the course of the disease from more disease-centered management to more palliative measures. 47 Currently, this shift is less obvious as even when lung function declines, the effect of the antifibrotic drugs may still be present. 93 Furthermore, these drugs may prevent the development of acute exacerbation12,94–96 and thereby protect the patient from a sudden decline in HRQOL or even death. In clinical practice, reassessing patients’ wishes and expectations, as well as incorporating the balance between efficacy and the burden of side effects, will guide decisions on discontinuation of antifibrotic treatments.
End-of-life care
Despite the fact that IPF has a prognosis worse than most malignancies, 97 end-of-life care is far less developed in this area of medicine than in oncology. Experiences from oncology have taught us that early palliative care improves the QOL and the mood of patients with metastatic lung cancer, and this, in turn, has resulted in less aggressive care and better survival. 98 There are a few issues that complicate end-of-life care in IPF. The disease is rare and relatively unknown to patients and the community. In qualitative interviews, patients and their relatives frequently rated their situation worse than patients with cancer. They perceived that in cancer everybody understands the seriousness of the disease and patients with cancer have ‘help coming from every direction’. 17 Another factor is the variable course of disease in IPF, in which sudden acute exacerbations can occur and prognosis is often unpredictable. Additionally, the timing of discussing end-of-life issues is difficult and should be tailored to the patients’ needs and wishes. There can also be dyssynchrony between patient and partners in wishing for information on this issue. Interactive questioning of patients in the Netherlands and Germany established that the majority of patients and partners prefer talking about the end-of-life early in the disease course, though we acknowledge that cultural differences may exist. 48 It is useful, however, to realize that discussing end-of-life may be anxiety provoking and can have a negative impact on HRQOL. 54 Sampson and colleagues found that patients with IPF clearly understand their prognosis but struggle to understand how their disease will progress. 17 Explaining the course of the disease, what to expect in the last phase, and palliative options may enable patients and families to make decisions in line with their values. In a recent study, only 13.7% of patients were referred to palliative care services, 99 indicating that the use of palliative care teams is underutilized at the moment. Alternatively, Bajwah and colleagues showed that interdisciplinary community care conferences improved symptoms and QOL. 100
Data from patients with other terminal diseases suggest that the majority of patients would prefer to die at home. 101 Lindell and colleagues showed that in a US cohort of patients, more than half of the patients died in hospital, a third on the ICU, and the remaining patients died in a hospice. 99 European studies showed that more patients died at home compared with the US study, but still a majority died in the hospital. 102 Hospital admission for respiratory-related causes in IPF is associated with high in-hospital mortality. 103 The limited options and devastating outcomes of hospital admission in the end-stage of the disease should be discussed with patients in an early phase to enable them to make decisions on limitations of care and allow them to choose the place of dying. At the patient’s request – if this is legally possible in the country where the patient is seeking treatment – different options of dying should also be discussed. Currently, worldwide, euthanasia has been legalized under strict conditions in a few countries. 104 In these countries, patients should be able to receive information on euthanasia. It is important to also discuss a ‘do not resuscitate’ code and a ‘do not intubate’ code with patients and family to avoid medical futility or unwanted interventions.
Conclusion
In a relentless disease such as IPF, striving to optimize HRQOL should complement the endeavour to prolong life. As symptoms, perceptions and reactions interact, and may change over time, a synchronized comprehensive management strategy is vital to match patients’ needs throughout the disease course. To do so, we propose the ABCDE of IPF care:
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
MM, JG, NT declare that they have no conflicts of interest. MW has received speaker fees, advisory board fees, and unrestricted research grants from Intermune, Hoffman la Roche, and Boehringer Ingelheim outside the submitted work. All fees were paid to her institution.