
Abstract
Pulmonary arterial hypertension (PAH) is a rare but fatal disease characterized by progressive vascular remodeling, which results in increased pulmonary vascular resistance and elevated pulmonary arterial pressure. These changes are detrimental to the right ventricle (RV). If not treated, it can eventually lead to maladaptive RV structural changes, right heart failure, and death. Late diagnosis at an advanced stage remains a significant issue that limits the effectiveness of existing treatments. PAH pathophysiology is mediated by several molecular pathways that act on different cell types, including endothelial cells, smooth muscle cells, and fibroblasts. These cells exhibit cancer-like properties, including increased proliferation, resistance to apoptosis, and metabolic reprogramming. This review provides new insights into clinical and diagnostic research on PAH. Herein, we discuss classification systems, their relevance and significance in PAH, innovative imaging techniques, and genetic testing to identify hereditary risk factors. The potential of artificial intelligence to improve disease detection and management is also discussed in the context of diagnostic workflows. Overall, we aim to provide new insights in this review and emphasize the critical need for early diagnosis, personalized treatment strategies, and continued innovation in PAH care to improve patient outcomes and quality of life.
Plain language summary
Pulmonary arterial hypertension (PAH) is a rare but life-threatening disease that causes progressive narrowing of the lung’s blood vessels. This increases resistance to blood flow, raises pressure in the pulmonary arteries, and puts significant strain on the right side of the heart. If left untreated, PAH can lead to heart failure and death. A major challenge in PAH care is late diagnosis, which reduces the effectiveness of available treatments. PAH develops through multiple molecular pathways affecting different cell types in the lungs, including endothelial cells, smooth muscle cells, and fibroblasts. These cells behave similarly to cancer cells, showing uncontrolled growth, resistance to cell death, and altered metabolism. However, despite advances in understanding PAH, the exact mechanisms that drive disease progression remain unclear, limiting the development of more effective treatments. This review highlights recent advances in PAH diagnosis and management, including improved classification systems, innovative imaging techniques, and genetic testing to identify hereditary risk factors. The emerging role of artificial intelligence in detecting PAH earlier and improving treatment decisions is also discussed. However, critical gaps remain, such as the lack of reliable biomarkers for early diagnosis and the need for personalized treatment approaches.
Keywords
Introduction
Pulmonary hypertension (PH) is a complex and life-threatening disorder characterized by elevated pressure in the pulmonary circulation, primarily resulting from the progressive vascular remodeling of small pulmonary arteries.1,2 Histopathological changes affecting all three arterial layers, including fibrosis, medial hypertrophy, and intimal proliferation, lead to increased pulmonary vascular resistance (PVR). 3 These structural alterations place chronic pressure overload on the right ventricle (RV), which can ultimately lead to right heart failure and death if left untreated. 4 RV function and the interaction between pulmonary vascular dysfunction and RV compensation are key prognostic determinants in patients with PH. 4 PH affects individuals across all age groups and presents with considerable clinical and hemodynamic variability. 5 However, the demographics and characteristics of patients diagnosed with PH have changed in the modern era.6–8 With more patients diagnosed with PH above the age of 60 years, exhibiting increased comorbidities such as diabetes, ischemic heart disease, and kidney dysfunction, recent reports from worldwide PH registries reveal a growing prevalence of the disease in the elderly. 8 Over the past few decades, the definition and classification of PH have evolved in response to advances in our understanding of its pathophysiology 9 (Figure 1). Group 1 PH was traditionally defined by the hemodynamic definition: mean pulmonary arterial pressure (mPAP) ⩾ 25 mmHg, pulmonary arterial wedge pressure (PAWP) ⩽ 15 mmHg, and PVR > 3 Wood units (WU). 10 The 2022 guidelines from the European Society of Cardiology (ESC) and European Respiratory Society (ERS) define PH as a mPAP ⩾ 20 mmHg at rest and PVR > 2 WU, as measured by right heart catheterization.11–13 Complementary hemodynamic measurements, including PAWP and PVR, help differentiate between the precapillary and postcapillary forms of the disease. While the World Health Organization (WHO) classification provides a broad etiological framework by organizing PH into five groups, 13 the ESC/ERS guidelines offer a more granular, risk-based approach. This risk stratification model incorporates clinical, functional, imaging, and hemodynamic parameters to guide treatment decisions and monitor disease progression. Group 1 PH or pulmonary arterial hypertension (PAH) includes idiopathic and heritable forms, commonly associated with BMPR2 mutations, as well as cases linked to connective tissue diseases, HIV infection, portal hypertension, congenital heart disease, and schistosomiasis. Notably, PAH is more prevalent in women, who paradoxically demonstrate better survival rates, a phenomenon often referred to as the “sex paradox.” 14 Recent epidemiological studies indicate that sex influences the development of PAH, with women being affected approximately four times more often than men, although this ratio can vary based on the disease subtype. 5

Updated clinical classification of pulmonary hypertension (PH). Five major diagnostic groups are commonly recognized based on etiology and hemodynamic criteria: (1) Pulmonary arterial hypertension (PAH), (2) PH due to left-sided heart disease, (3) PH due to lung diseases and/or hypoxia, (4) PH due to pulmonary artery obstructions, and (5) PH with multifactorial or unclear mechanisms. The relative prevalence of each group is visually depicted, emphasizing the rarity of PAH and the higher frequency of PH associated with left-sided heart disease.
Despite advances in targeted therapies, PAH remains a progressive disorder that is associated with substantial morbidity and mortality. Delays in diagnosis, often extending beyond 2 years due to the insidious onset of nonspecific symptoms, continue to hinder early therapeutic intervention and contribute to poor outcomes. In this review, we provide a comprehensive update on recent developments in the classification, diagnosis, and management of PAH, with a particular focus on recent translational and clinical advances. We discuss the evolving clinical classification of PAH and its implications for individualized care, highlight the emerging role of genetic testing, multi-omics, and computational modeling in refining risk stratification and informing therapeutic strategies, and examine the integration of artificial intelligence and advanced imaging techniques into diagnostic workflows.
Vascular remodeling and cellular heterogeneity
PAH pathogenesis
The pathogenesis of PAH is characterized by progressive narrowing and stiffening of the pulmonary vasculature, leading to increased PVR and RV afterload (Figure 2). These changes are driven by alterations in multiple cell types in the distal pulmonary arteries, including endothelial cells, smooth muscle cells, fibroblasts, and inflammatory cells. The endothelium serves as a key sensor of pathogenic stimuli, such as hypoxia, shear stress, and inflammation, regulating vascular tone and homeostasis. 15 Endothelial dysfunction in PAH disrupts the balance between vasoconstrictors and vasodilators and alters pro- and antiproliferative, thrombotic, and inflammatory signaling.16–19 Histopathological changes include fibrosis, neointimal hyperplasia, and inflammatory infiltration, primarily driven by abnormal vascular cell proliferation, resistance to apoptosis, and extracellular matrix remodeling.20,21 These processes are further promoted by hypoxia, impaired vasodilation, excessive vasoconstriction, and perivascular inflammation. The resulting vascular dysfunction places chronic stress on the RV, leading to compensatory hypertrophy and dilation.22,23 Over time, these adaptations fail, resulting in RV dysfunction, heart failure, and increased mortality (Figure 2). The precise mechanisms underlying these transitions remain unclear, though.

Cellular and pathophysiological drivers of vascular remodeling and right heart failure in PAH. Vascular remodeling in distal pulmonary arteries, mediated by endothelial cells, smooth muscle cells, fibroblasts, and inflammatory cells, leads to intimal proliferation, medial hypertrophy, adventitial fibrosis, and perivascular inflammation. These changes increase pulmonary vascular resistance, imposing chronic pressure overload on the right ventricle. In response, cardiomyocytes, fibroblasts, and inflammatory cells contribute to right ventricular hypertrophy, interstitial fibrosis, inflammatory infiltration, and progressive contractile dysfunction. The figure illustrates the cascade and progression from pulmonary vascular alterations and increased afterload to right ventricular compensation and, ultimately, decompensation.
Structural and cellular heterogeneity
Over the past decade, significant advancements have been made in our understanding of the pathobiology of PH. Researchers have attempted to elucidate the structural composition of PH-affected tissues and have developed various in vitro, in silico, and in vivo models to better understand and characterize the disease and identify new therapeutic targets. 24 These efforts have significantly improved our understanding of the molecular basis of PH and the structural changes affecting the heart and lung vasculature. Technological advances have provided valuable structural insights. Notably, synchrotron micro-CT has identified four distinct types of lesions in PH, including type 1 branched lesions, which are complex and expansive. 25 Type 2 lesions indicate shunts between the pulmonary arteries and systemic circulation via the bronchial vessels. Type 3 destructive lesions extend from the pulmonary arteries to the pulmonary veins, with obstructive lesions that eventually recanalize. These findings are currently being integrated with traditional histological techniques and spatial transcriptomic methods to better understand the disease morphology and the underlying dysregulation of gene expression. 26 Recent studies have also used sectional analysis of the pulmonary arteries from distal to proximal segments, coupled with single-cell sequencing, to discern structural alterations in vascular and immune cell populations. 26 Researchers have identified four distinct types of smooth muscle cells: traditional contractile, synthetic, oxygen-sensing, and carbon blast phenotypes. Transcriptional changes, notably in synthetic and contractile cells, suggest potential transitions between fibroblasts and smooth muscle cells, with the synthetic type predominantly found in the neointima of lesions. Moreover, digital spatial transcriptomics have expanded these findings beyond plexiform lesions, revealing unique transcriptional signatures in obstructive and adventitial lesions and identifying markers such as IGFBP4 in plexiform lesions and Endoglin and Complement Factor D in the adventitia. 26
Clinical overview and diagnosis of PH
The classification of PH has undergone significant refinement, reflecting an enhanced understanding of its diverse etiologies and clinical needs. Earlier frameworks, such as the WHO system, relied primarily on shared pathophysiological features. Current practice is guided by the 2022 ESC/ERS guidelines, which incorporate more detailed clinical, imaging, and hemodynamic criteria.11–13 Refinements in classification have also accounted for patients with overlapping phenotypes, such as those with chronic lung disease or multiple comorbidities, enabling more nuanced and individualized management strategies.11–13 Group 1 PH or PAH remains a particularly aggressive form of the disease and a primary focus of diagnostic and therapeutic research. Treatment options have expanded considerably over the past few decades. However, PAH is still associated with a poor prognosis. Historical data indicate a median survival of 2.8 years from diagnosis, with 1-, 3-, and 5-year survival rates of approximately 68%, 48%, and 34%, respectively. 27 Current registry data suggest a prevalence of 15–52 cases per million adults in the United States. 28 However, survival rates have not improved significantly in recent years, likely due to persistent diagnostic delays and the evolving demographics of newly diagnosed patients.
Timely diagnosis of PAH is critical but remains a challenge, largely because its symptoms are nonspecific and often resemble those of more common respiratory and cardiovascular conditions 12 (Figure 3). Patients typically present with dyspnea on exertion, fatigue, chest discomfort, dizziness, palpitations, and peripheral edema. 12 These symptoms frequently lead to misdiagnosis or delayed referral for appropriate evaluations. Diagnostic workup begins with a comprehensive clinical assessment and proceeds through a structured algorithm designed to exclude secondary causes and confirm the diagnosis. 12 Functional capacity is commonly assessed using the six-minute walk test, while noninvasive imaging tools, such as echocardiography and computed tomography, provide valuable insights into cardiac morphology and pulmonary artery anatomy. Right heart catheterization remains the gold standard for confirming PH and determining its hemodynamic classification. Laboratory evaluations, including hematological and biochemical tests, help identify contributing systemic disorders and organ dysfunction. Monitoring hemodynamic parameters, such as mPAP, PVR, PAWP, cardiac output, and right atrial pressure, is essential for determining disease severity and guiding patient management (Figure 4). As diagnostic strategies have become more sophisticated, early recognition and risk stratification have improved. Nonetheless, persistent delays and under-recognition in clinical settings underscore the need for ongoing innovation in diagnostic approaches and broader awareness of PAH’s subtle early manifestations of the disease.
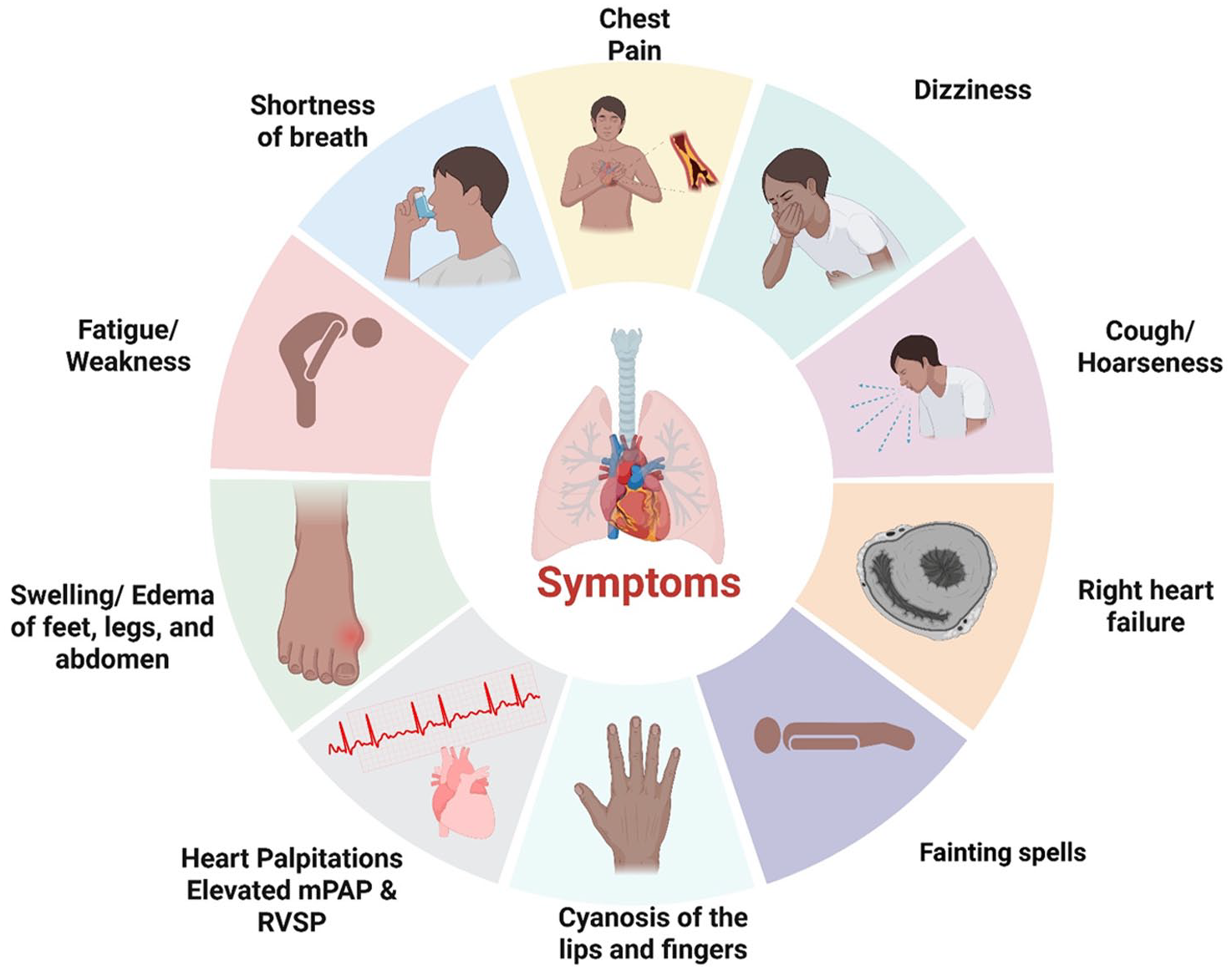
Symptoms of pulmonary arterial hypertension. Common PAH symptoms may vary by severity and type. These symptoms include shortness of breath that worsens with exertion or at rest; chest pain or tightness during activity or at rest; fatigue; swelling in the legs, ankles, or feet (peripheral edema); dizziness or lightheadedness, potentially leading to fainting; rapid heartbeat (palpitations); bluish skin or lips (cyanosis), indicating low blood oxygen; dry cough; and weight gain due to fluid retention.

Traditional diagnostic techniques in PAH. Key traditional methods for PAH assessment include the 6-minute walk test for functional capacity, right heart catheterization for definitive hemodynamic diagnosis, and ECG for detecting right heart strain and conduction abnormalities. Echocardiography offers a noninvasive evaluation of right ventricular structure and function and helps identify underlying causes. Routine blood tests support the assessment of cardiac strain, organ failure, and comorbidities.
Advancements in imaging and diagnostics
Advancements in molecular diagnostics
Further insights were provided by a Stanford study that used multiplex ion-beam imaging or time-of-flight techniques to characterize cellular populations within pathological lesions using panels of up to 96 specific antibodies. 29 This approach has pinpointed immune subsets, including a unique enrichment of eosinophils in lesions linked to mutations in bone morphogenetic protein receptor type 2 (BMPR2) in patients with PH. 29 Other studies have integrated genome-wide association study (GWAS) data, clinical observations, and Bayesian regulatory networks using a large biobank. 30 Interestingly, they identified a gene co-expression cluster associated with severe PH, but paradoxically linked it to a decreased risk, suggesting a phenotype of compensated severe PH. 30 This cluster included both known and novel genetic targets, with one new gene identified as a potential exacerbator of PH. The authors experimentally confirmed its role in the regression of PH lesions, underscoring the value of multiomic approaches and biobanks. The need for new biomarkers and innovative imaging techniques stems from the lack of disease-specific symptoms and late-stage diagnosis. Common markers, such as BNP or NT-proBNP, which are indicative of cardiac stress, can be influenced by variables such as fluid volume changes, left heart failure, and variations in patient demographics, such as sex and obesity, which diminish their specificity for PH. 31 In routine clinical assessments, the six MWT is a straightforward method that is often not adjusted for age, height, or weight and can be affected by minor factors. 32
Innovative imaging techniques
Advanced imaging techniques, such as cardiac magnetic resonance and echocardiography, are crucial for PAH screening. However, they can only offer indirect insights into PVR, as they often fail to precisely assess cardiac pressure, particularly by echocardiography. Right heart catheterization remains the gold standard for diagnosing PAH, despite its invasiveness and typical restriction to resting measurements.33–35 There is an increasing shift toward performing invasive cardiopulmonary exercise testing to increase diagnostic accuracy, which requires further studies to determine concurrent conditions such as lung diseases. 36 Despite these established methods, our understanding of pulmonary vascular disease relies heavily on indirect assessment of vascular resistance rather than direct observation of the disease process. Emerging techniques for evaluating pulmonary circulation and right heart function include the analysis of noncontrast chest computed tomography (CT) scans, which allow for a detailed examination of small and large blood vessels. 37 This analysis allows for the quantification of vascular pruning and enhances functional respiratory imaging used to assess changes in vessel wall thickness in response to inhaled nitric oxide or other therapeutic agents such as sildenafil. 38 For example, improvements in peripheral small-vessel structures after therapy illustrate the potential of these novel diagnostic approaches to transform our understanding and management of PAH. 32
Quantitative assessment of vascular structures correlates closely with invasive measures of pulmonary hemodynamics, providing valuable insights into disease severity and treatment response in PAH. Advanced technologies, such as photon counting and dual-energy CT, now enable high-resolution imaging, improving the diagnosis of chronic vascular obstructions and perfusion deficits.39,40 Reduced signals on iodine perfusion maps allow for a comprehensive evaluation of vessel integrity and downstream perfusion. In CTEPH, four-dimensional flow MRI quantifies parameters such as vortex duration, helicity, and vorticity, which are closely correlated with the mPAP and right ventricular function. Notably, a prolonged vortex duration in the main pulmonary artery is a highly sensitive and specific marker of elevated mPAP.41,42 Hyperpolarized xenon MRI further enhances disease characterization by allowing the visualization of gas exchange in the lungs. In PAH, this technique detects distinct red blood cell (RBC) signal abnormalities that correlate with impaired gas transfer,43–45 which differ from the patterns observed in chronic obstructive pulmonary disease or idiopathic pulmonary fibrosis. 46 These cutting-edge imaging modalities deepen our understanding of PAH pathophysiology and facilitate more precise diagnosis and management, bridging clinical practice and technological innovation. In patients with PAH and left heart failure, airspace and membrane signals on hyperpolarized xenon MRI are typically preserved, whereas RBC signals are markedly abnormal, reflecting impaired gas transfer to red blood cells. 34 Temporal changes in these signals can differentiate various forms of PH associated with left heart disease and can be mathematically modeled to yield additional physiological insights, such as correlations with the capillary blood volume. Notably, this technology is sensitive to therapeutic interventions; for example, inhaled treprostinil has been shown to improve RBC signals within minutes.
The integration of lung and cardiac imaging is essential for distinguishing PAH from other causes of PH, such as pulmonary venous hypertension.11,47,48 Echocardiography remains a key tool in this context, while advanced imaging modalities can reveal specific features, such as mitral valve remodeling in connective tissue disease or congenital anomalies in patients with heart disease, which may not be apparent through standard approaches. This integrated imaging strategy supports a more accurate diagnosis and personalized management of complex cardiopulmonary disorders, highlighting the transformative potential of advanced imaging in clinical practice (Figure 5).

Advanced diagnostic techniques in PAH. Advanced diagnostics provide greater precision in PAH evaluation. Cardiac MRI and 4D flow MRI allow detailed analysis of right ventricular structure, function, and hemodynamics. Noncontrast and dual-energy computed tomography (CT) visualize vascular and structural changes. Genetic testing enables the identification of heritable forms of PAH. AI/ML-driven algorithms and integrative omics approaches enhance early diagnosis, risk prediction, and individualized patient profiling.
Standardizing advanced imaging in clinical practice and research
Diverse diagnostic insights from echocardiography are crucial for managing patients with complex cardiac conditions. Novel techniques, such as RV strain measurement during exercise and cardiac imaging approaches, may offer comprehensive insights. Specifically, the ability of cardiac MRI to evaluate RV function via 4D flow imaging proves its potential use as a comprehensive diagnostic tool with high-resolution imaging capabilities for cardiac structures.49,50 For example, patients with CTEPH often exhibit markedly enlarged right ventricles and clear perfusion abnormalities, evidenced by reduced perfusion in the right lower lobe, which is consistent with findings from angiography. 51 Similarly, significant enlargement of the left atrium was observed in patients with pulmonary venous hypertension. These imaging modalities provide a holistic view of cardiac and pulmonary circulation, offering a robust framework for accurate diagnosis and tailored treatment strategies.52,53 While these imaging tools have the potential to serve as endpoints in clinical trials, thereby enhancing the efficacy and precision of clinical research, standardization is necessary to ensure their uniform application across different settings. 54
Risk stratification and patient-centered management
In clinical practice, determining an appropriate therapeutic strategy for patients involves a risk stratification approach.55,56 This approach assesses the mortality risk of a patient within the following year using scoring systems such as the REVEAL risk score calculator and the ESC/ERS risk assessment models. High-risk patients may initially require aggressive intervention with a combination of three drugs, whereas those at a lower risk may benefit from a more gradual treatment approach. 57 Thus, the primary factors considered in all scoring systems included the patient’s functional class, walking distance, and biomarkers such as BNP, along with other clinical and biological parameters. 57 This approach ensures tailored therapy that aligns with the severity and specific characteristics of a patient’s condition. Dynamic imaging plays a crucial role; however, decisions regarding therapy initiation, whether to start with aggressive triple combination therapy, including parenteral cycling plus a combination regimen, or opt solely for oral therapy, are guided only by the severity scoring system.55,57 This system facilitates initial and detailed assessments to determine whether a patient is at high risk and requires aggressive management. For non-high-risk patients, a sequential treatment approach is recommended, starting with a basic combination and adjustment as needed. The variability within the scoring system helps determine whether a patient fits a low-risk profile or, on the contrary, whether the treatment goals necessitate intensified therapy.55,57 Another critical aspect is the management of comorbidities, particularly in older patients. A patient-oriented approach is essential, as comorbidities significantly influence treatment strategies and efficacy. 58
Genetic insights and testing in PAH
PAH is a genetically heterogeneous disorder, with both inherited and de novo mutations contributing to disease susceptibility, onset, and progression. The discovery of pathogenic variants, particularly in genes related to the transforming growth factor-beta (TGF-β)/bone morphogenetic protein (BMP) signaling axis, has reshaped our understanding of PAH pathobiology. Genetic testing has emerged as a critical component of modern PAH management, offering diagnostic clarification in idiopathic cases, stratifying risk, guiding surveillance of asymptomatic carriers, and informing reproductive and therapeutic decision-making.
Importance of genetic testing
Genetic testing has become an indispensable tool for the diagnosis, risk stratification, and clinical management of PAH, particularly in idiopathic and heritable forms. Among the known genetic contributors, mutations are the most frequently identified, present in approximately 70%–80% of heritable PAH cases and up to 25% of idiopathic PAH. 59 These heterozygous mutations typically result in haploinsufficiency and disrupt BMP signaling, leading to aberrant vascular cell proliferation, inflammation, and resistance to apoptosis, which are all hallmarks of PAH pathobiology. 59 Preclinical efforts targeting the restoration of BMPR2 signaling, such as ligand traps (e.g., sotatercept) and gene therapy, are currently under investigation. Data from a meta-analysis of 1550 patients with PAH (UK National Cohort) indicated that BMPR2 mutations were present in approximately 29% of cases. 60 Carriers of these mutations are diagnosed at a younger average age (35.4 years) than noncarriers (42.0 years). 60 At diagnosis, BMPR2 mutation carriers exhibit more severe hemodynamic parameters, including higher mPAP, elevated PVR, and lower cardiac index.60,61 In addition, only 3% of mutation carriers respond to acute vasodilator testing compared to 16% of noncarriers. 60 Survival analysis revealed that BMPR2 mutations are associated with an increased risk of death, lung transplantation, and all-cause mortality, with the impact being more pronounced in patients diagnosed before the age of 30. 60 These findings underscore the importance of genetic testing for BMPR2 mutations in patients with PAH, as the mutation status provides valuable prognostic information and can guide clinical management strategies.
A systematic screening for PAH in adults carrying a BMPR2 mutation demonstrated that asymptomatic BMPR2 mutation carriers have a significant risk of developing incident PAH, ranging from 0.99% per year in males to 3.5% per year in females.62,63 As such, the 2015 ESC/ERS guidelines recommend annual echocardiography for individuals with known PAH-associated mutations and first-degree relatives of affected individuals (Class IIb, Level C). Mutation identification can also inform reproductive decision-making, including the consideration of prenatal or preimplantation genetic diagnosis, particularly given the severity and incomplete penetrance of the disease. In the phase II PULSAR trial, 76 patients with PAH were genotyped for variants in PAH-associated genes (including BMPR2, ACVRL1, CAV1, EIF2AK4, ENG, KCNK3, KCNA3, and SMAD9) to assess whether genetic status influences the response to sotatercept. 64 Pathogenic variants were identified in 25 patients (23 with BMPR2), who were younger and more often on triple therapy but had less severe clinical features at baseline than noncarriers. 64 After 24 weeks of treatment, changes in PVR and 6-min walk distance were similar across the variant and non-variant groups. 64 BMPR2 mRNA expression and NT-proBNP levels also showed no genotype-specific differences in response. 64 The adverse events were consistent with those reported previously. These findings indicate that the efficacy and safety of sotatercept are not significantly affected by BMPR2 mutation status, supporting its use across genetically diverse PAH populations.
Genetic counseling and family implications are also important considerations
Genetic testing and counseling have become increasingly important in PAH management. Approximately 25%–30% of patients with idiopathic PAH have a Mendelian genetic cause for their disease. 59 Both patients and clinicians show strong interest in understanding the genetic underpinnings of PAH,65,66 as testing not only guides individual management but also enables the identification of at-risk relatives, supporting more informed clinical decisions and improved long-term outcomes. 67 Genetic evaluation is particularly recommended for adults with idiopathic or heritable PAH, pulmonary veno-occlusive disease, and those with relevant family histories or risk exposures (e.g., congenital heart disease and anorectic use), as well as for pediatric patients with developmental lung disorders or congenital diaphragmatic hernia. 68 Testing symptomatic individuals is generally straightforward. Clinicians with basic genetics training can initiate testing using targeted gene panels for established PAH genes, with escalation to whole-exome or genome sequencing if indicated. This streamlined approach has made genetic assessment more accessible in routine care. Advancements in genomic technologies have expanded our understanding of PAH genetics, enabling the discovery of new disease-associated genes through unbiased genome-wide approaches. Importantly, the detection of a dominantly inherited mutation (e.g., BMPR2 and ACVRL1) has direct implications for cascade testing in first-degree relatives, whereas the identification of a recessive variant (e.g., EIF2AK4) informs recurrence risks for siblings and future offspring.59,69,70 For asymptomatic family members, a negative test for a known familial mutation lowers their risk to that of the general population.BMPR2 mutations, present in roughly 29% of PAH patients, are associated with younger age at diagnosis (35.4 vs 42.0 years), more severe hemodynamic compromise, and reduced vasoreactivity. Notably, the PULSAR trial demonstrated that sotatercept is effective regardless of BMPR2 status, supporting its use across various genetic backgrounds.
To maximize clinical utility, genetic counseling is essential. Patients and families must be educated on test interpretation, incomplete penetrance, variable expressivity, reproductive implications, and protections against genetic discrimination. Ideally, counseling should be delivered by trained geneticists or PAH specialists with relevant expertise. However, in resource-limited settings, clinicians equipped with basic genetics training and access to centralized or virtual counseling services can effectively support patients throughout the testing process. The initial evaluation typically involves testing a targeted panel of known PAH-associated genes. If no pathogenic variant is identified but clinical suspicion remains high, further sequencing using whole-exome or whole-genome approaches may be appropriate. Testing should be conducted in accredited laboratories to ensure analytical rigor and accurate variant interpretation. Practices vary globally regarding disclosure of research-derived variants, underscoring the importance of informed consent and clear pretest education. Pediatric PAH warrants special consideration because of its distinct genetic profile compared with adult-onset disease. Children with PAH are more likely to carry rare inherited variants, including those in TBX4, SOX17, and EIF2AK4, and often present with developmental abnormalities. Therefore, genetic testing and counseling should be routinely offered in pediatric cases to guide diagnosis, prognostication, and family planning. Overall, systematic genetic testing should be considered in all patients with idiopathic, familial, early onset, symptomatic, or asymptomatic PAH with a family history of PAH, as well as in those with suspected pulmonary veno-occlusive disease or pulmonary capillary hemangiomatosis. Incorporating genetics into routine clinical practice improves diagnostic precision, enables early detection in at-risk relatives, and informs long-term surveillance and treatment strategies. Continued investment in clinician education, counseling infrastructure, and laboratory quality assurance is essential to realize the full clinical impact of genetic testing in PAH.
Advances and future directions in omics
Advancements in omics technologies have significantly enhanced our understanding of the molecular risk factors in PAH, enabling presymptomatic monitoring and timely intervention in mutation carriers.71,72 Multiomic studies integrate diverse platforms, such as genomics, proteomics, and metabolomics, to comprehensively characterize biological systems, identify biomarkers, and inform personalized medicine, drug development, and disease classification.73,74 By generating individual molecular signatures, omics approaches can detect early disease markers and guide treatment. For example, GWASs have identified risk variants associated with PAH.30,71 The combination of public multiomics datasets further facilitates the discovery of genetic causes and novel therapeutic targets. As these technologies advance, multiomics strategies are expected to refine PH classification and improve patient outcomes.
Integration of artificial intelligence
Despite recent therapeutic advancements, PAH remains a life-threatening condition with high morbidity and mortality rates. Its growing clinical heterogeneity, driven by comorbidities, variable treatment responses, and diverse molecular profiles, continues to limit the effectiveness of standard therapy. Precision medicine offers a promising alternative by tailoring interventions to individual patient characteristics, including genetic, environmental, and lifestyle factors. To apply this approach in practice, clinicians require tools that can process, integrate, and interpret complex data from multiple sources. Artificial intelligence (AI) meets this need by integrating information from advanced imaging, genetic testing, and clinical biomarker studies. Recently, AI has shown promise in enhancing diagnostic accuracy, improving risk prediction, and supporting individualized treatment decisions, thereby advancing the integration of precision medicine into routine clinical practice.
Opportunities for AI in PAH
In clinical settings, AI-powered platforms demonstrate performance comparable to expert interpretations. For instance, deep learning algorithms trained on chest radiographs enable the early identification and referral of at-risk patients. 75 Similarly, AI models applied to high-resolution CT quantify parenchymal and vascular alterations, supporting refined disease phenotyping across PH subtypes. 76 Beyond imaging, machine learning (ML) techniques are increasingly being applied to high-dimensional clinical, molecular, and hemodynamic datasets. 77 These approaches surpass traditional tools in terms of early disease detection, phenotype classification, and risk stratification. Notably, predictive ML classifiers can predict PAH onset months before clinical diagnosis, opening a window for preemptive intervention. 78 ML-driven triage systems are also being evaluated to guide right heart catheterization in cases with ambiguous diagnoses, potentially expanding access to definitive hemodynamic assessments. 79 Meta-analyses support the incorporation of ML-enhanced modalities, including electrocardiography, echocardiography, and advanced imaging, into the routine evaluation of PAH. 77 Computational frameworks incorporating comorbidities, pharmacogenomics, and sex-specific responses may accelerate in silico simulations, refine preclinical testing, and inform clinical outcomes.
Challenges and limitations
While the integration of AI may improve early diagnosis, the validation and generalizability of predictive models remain key concerns. Many algorithms have been developed using retrospective or single-institution datasets, which can limit their applicability across broader and more heterogeneous patient populations. 80 The current absence of standardized development pipelines, along with variability in data quality from imaging and information available from electronic health records, further undermines reproducibility.81,82 As a result, the interpretability of many AI models remains limited, hindering the seamless integration of AI into routine practice. Similarly, the field of omics faces substantial technical and translational challenges. The complexity and high dimensionality of omics data demand rigorous statistical and bioinformatic approaches to ensure valid and reproducible findings. 83 Reproducibility remains a challenge when studies are based on limited cohorts or lack appropriate controls. Moreover, the cost and infrastructure required for comprehensive omics profiling limit its widespread clinical adoption, especially in resource-constrained settings.
Conclusion
Recent advances in diagnostics and therapeutics have significantly improved PAH management. Nevertheless, major challenges persist, including delayed diagnosis, substantial patient heterogeneity, and variable treatment response. Emerging tools, such as next-generation imaging, genetic screening, multiomic profiling, and artificial intelligence, offer unprecedented opportunities to refine diagnostic accuracy, risk stratification, and personalized treatment strategies. However, the widespread adoption of these innovations requires standardized validation protocols and seamless integration into clinical practice. Ultimately, strong collaboration among molecular biologists, clinicians, imaging specialists, and computational scientists will be critical to translate these innovations into meaningful clinical benefits for patients with PAH.