
Abstract
Background:
Joubert syndrome (JS) is an autosomal recessive disorder with a distinctive mid-hindbrain malformation known as the “molar tooth sign” which involves the breathing control center and its connections with other structures. Literature has reported significant respiratory abnormalities which included hyperpnea interspersed with apneic episodes during wakefulness. Larger-scale studies looking at polysomnographic findings or subjective reports of sleep problems in this population have not yet been published.
Objectives:
The primary objectives were (1) compare a large group of children with JS and their unaffected siblings for caregiver-reported sleep difficulties. Secondary objectives were (1) present new polysomnography (PSG) data on our JS cohort; (2) review sleep disordered breathing (SDB) in other rare congenital hindbrain anatomic abnormalities.
Design:
We conducted a cross-sectional study on a cohort of 109 families affected by JS.
Methods:
Pediatric Sleep Questionnaire (PSQ) and the Children’s Sleep Habits Questionnaire (CSHQ) along with general medical health information focused on respiratory and sleep problems were mailed to all patients and families. Caregivers were asked to complete the survey for both children with JS and unaffected siblings, if any. Baseline diagnostic PSG was retrospectively reviewed for those with available studies, and the sleep parameters were compared to a referent cohort.
Results:
Study participants with JS were older than their unaffected siblings (p = 0.02). Genetic mutations were available for 41 out of 118 individuals, with the most common mutation being MKS3 (31.4%). Patients with JS had higher scores in the PSQ compared to their unaffected siblings (p < 0.001). PSG data showed severe SDB with apnea-hypopnea index (AHI) of 23 ± 15 events/h in patients with JS. Events were primarily obstructive (obstructive AHI 18 ± 15 events/h vs central AHI 4 ± 4 events/h). Abnormal sleep architecture with increased arousal indices, decreased efficiency, and more time awake and in light sleep or wakefulness when compared to the referent data.
Conclusion:
SDB is common and severe in patients with JS, and the significantly greater obstructive component reported in this cohort makes it necessary to perform complete PSG studies to address or prevent clinical manifestations in this at-risk population. PSQ could represent a viable method to screen for SDB in JS.
Background
The anatomic respiratory control center is canonically regarded to be within the medulla. Indeed, injury to this region results in profound dysregulation of breathing control, most often manifesting as central apneas and hypoventilation most pronounced during sleep. However, the respiratory control network extends well beyond the physical boundaries of the brainstem, as evidenced by individuals with clinical conditions involving hindbrain structures outside of isolated brainstem abnormalities. In this article, we will present new data on pediatric patients diagnosed with Joubert syndrome (JS) and their associated sleep problems. Additionally, we will review sleep disordered breathing (SDB) in other rare congenital hindbrain anatomic abnormalities.
JS is an autosomal recessive disorder clinically characterized by hypotonia, ataxia, abnormal eye movements, and intellectual disability, identifiable by a distinctive mid-hindbrain malformation known as the “molar tooth sign” (MTS) on magnetic resonance imaging (MRI). 1 Recent identification of genetic abnormalities in affected individuals has placed JS in a larger spectrum of phenotypic disorders united by ciliary dysfunction, or ciliopathy. 1 The prevalence of JS is estimated at 1:100000 in the US (JS and JS related disorders). 2 Other frequent clinical manifestations include irregular breathing, developmental delay, facial dysmorphism, macrocephaly, and head tilting. Of note, individuals with significant hypotonia are more vulnerable to dysphagia and pulmonary injuries caused by aspiration. Moreover, JS patients often experience progressive impairment in various bodily systems, such as the eyes, kidneys, liver, and musculoskeletal structures. 3 Additionally, behavioral problems such as hyperactivity, aggressive behavior, decreased verbal fluency, memory, and response are not uncommon in individuals in JS, 4 some of which overlap with problems seen in children with chronic sleep difficulties. 5
The initial reports by Joubert et al.6,7 describe significant respiratory abnormalities which included hyperpnea with extrapolated rates of up to 168 breaths per minute interspersed with apneic episodes lasting 5–12 s during wakefulness. Though respiratory abnormalities attenuated somewhat with time, they continued into adulthood. 8 In addition, sleep disturbances were reported in the original proband. 8 Since that time, further case reports in children and adults have illustrated significant sleep-related breathing abnormalities on polysomnography (PSG),9 –11 some severe enough to necessitate mechanical assisted ventilation.10,12 Findings included both central apnea as well as the hyperpnea–apnea pattern previously described in the original reports. One recent study queried a small sample of young adults/caregivers of children with JS, reporting that SDB on questionnaire was common in this population. 13 Additionally, another recent case report described clusters of paroxysmal motor episodes after arousals triggered by apneic events. 14 However, larger-scale studies looking at PSG findings or subjective reports of sleep problems in this population have not yet been published.
Given a recent explosion in evidence linking SDB with cognitive, behavioral, and general medical sequelae, 15 for which children with JS are already at risk, the characterization of sleep disorders, including SDB, is an important foundational step in efforts to offer novel treatments for this population. Should a link between the two be established in JS, this would provide a venue for possible early interventions and treatment to help better optimize developmental potential. Thus, our aim for this study was to systematically survey a large group of children with JS and their unaffected siblings for sleep difficulties using validated caregiver-reported pediatric sleep measures, as well as to provide an initial description of a cohort of children with JS who have undergone diagnostic PSG. Additionally, we will review SDB in other rare congenital hindbrain anatomic abnormalities.
Materials and methods
Study design, settings, and participants
We conducted a cross-sectional study on a cohort of 109 families affected by JS who are enrolled at the University of Washington (UW) Joubert Research Center between 2008 and 2010. Patients were referred to the UW by clinical collaborators throughout the world and the JS and Related Disorders Foundation. The procedures used in this study were in agreement with the research protocol approved by the Institutional Review Boards at the UW, and all participants or their legal representatives provided written informed consent. Inclusion criteria included clinical findings of JS (intellectual impairment, hypotonia, ataxia) and diagnostic or supportive brain imaging findings (MTS on MRI vs cerebellar vermis hypoplasia on computed tomography scan) or having a sibling with JS along with supportive clinical or imaging features. Exclusion criteria were families who did not consent to participating in the study.
Genetic and clinical information was obtained by direct examination, medical record review, family intake forms, and/or pedigree ascertainment.
Questionnaires
To ascertain current and consistent data, a structured patient questionnaire was mailed to all patients and their families for whom an address was available. This two-page survey focused on sleep and respiratory problems, and caregivers were asked to complete the survey for both children with JS and unaffected siblings (if any) at home. The survey incorporated the Pediatric Sleep Questionnaire (PSQ), 16 the Children’s Sleep Habits Questionnaire (CSHQ), 17 and questions about general medical health, including those focusing on respiratory/pulmonary problems, prior treatment, and diagnoses. Caregivers respond on a Likert scale for frequency of reported sleep habits, with higher scores indicating more disturbed sleep. Thirty-one items out of the original 33 were included in this study’s survey, with two questions regarding daytime sleepiness omitted. We recognize potential recall and performance bias given the nature of caregiver-administered questionnaires.
Polysomnography
In addition to the prospectively collected survey data, we retrospectively reviewed PSG which had been done in children with JS enrolled in the registry at Seattle Children’s Hospital. PSGs were obtained as part of clinically indicated care through the Sleep Disorders Center at Seattle Children’s Hospital; IRB approval was obtained to examine PSG data. Only baseline diagnostic sleep studies are included in this review. Each PSG lasted at least 6 h and was performed in an accredited pediatric-specific sleep laboratory setting, which included a private darkened room free of distraction with a parent or guardian present. The following physiologic parameters described in the American Academy of Sleep Medicine (AASM) version 2.6 scoring manual were monitored: electroencephalogram, electro-oculogram, submental and anterior tibialis electromyograms, electrocardiogram, oxygen saturation via pulse oximeter, and thoracic and abdominal movement. Other parameters not included in the AASM scoring manual, oronasal airflow measured by thermistor and pressure transducer, and transcutaneous and/or expired end-tidal carbon dioxide (summarized as mean CO2 in Table 6), were also monitored. Direct observation of sleep was done by an attendant technologist. All data were recorded into a computer-based acquisition and analysis program (Rembrandt®, Buffalo, NY, USA) and interpreted by a board-certified Sleep Medicine physician in accordance with pediatric practice parameters of the AASM. 18 The Apnea–Hypopnea Index (AHI) was defined as the total number of respiratory disturbances averaged per hour of total sleep time, and further divided into those caused by obstructive apneas/hypopneas and those caused by central events. The Oxygen Desaturation Index was defined as the total number of desaturations of at least 3% averaged per hour of total sleep time.
Data analysis
Results are primarily descriptive. Data from the survey were compared between the individuals with JS and unaffected siblings (controls) using the T-tests for continuous variables or chi-squared tests for categorical/ordinate variables. Logistic regression was used to correlate findings from the survey to objective PSG results in a small subset of participants. To reduce bias, patients with missing data were removed from the data analysis, missing data accounted for <5% of the study. Statistical comparison was not performed on PSG results between the JS group and referent population. Analyses were performed using STATA-10 (College Station, TX, USA).
Material and methods section follow the STROBE guidelines for cross-sectional studies. 19
Results
Demographics
Surveys were sent to 173 families, and 166 surveys were returned: 118 from individuals with JS and 48 from unaffected siblings, representing a total of 109 unique families. The mean age for patients with JS was 16.7 years (±7.9) and 13.9 years (±5.4) for unaffected siblings and parent’s subjective response to overweight status was 14% for JS and 8% for unaffected siblings, both variables were ns (see Table 1). Genetic mutations (Table 2) were available for 41 out of 118 JS patients, with the most common mutation being MKS3 (14 out of 41 patients). There was no data found regarding genetic testing for the rest of the JS patients.
Patient demographics.

Data was obtained from a yes or no answer provided by the families in the survey, which did not include a numerical value.
JS, Joubert syndrome.
Genetic mutations in the Joubert Syndrome cohort, n = 41.

Data available for 41 out of 118 JS patients. There was no data found regarding genetic testing for the rest.
JS, Joubert Syndrome.
Medical history
Questions about medical history were included in the survey. No statistical differences were observed between the group with JS compared with controls regarding current or previous ventilatory support or cardiopulmonary monitoring (Table 3). Rates of adenotonsillectomy, prior PSG, diagnosis of SDB, or family history of SDB were similar between groups.
Past medical history, results shown in percentages (p = ns).

Only two sibling sets.
JS, Joubert Syndrome; PSG, polysomnography; SDB, sleep disordered breathing.
Sleep questionnaires
There was no difference in mean CSHQ score between patients with JS and their unaffected sibling (35.7 ± 7.3 vs 34.4 ± 5.8, respectively). Among the 118 JS patients, 19 of them (16.1%) had a CSHQ > 41; and among the 48 unaffected siblings, 6 of them (12.5%) had a CSHQ > 41. There was also no difference when comparing the scores of those with CSHQ > 41 between JS patients and their unaffected siblings (48.9 ± 7.2 vs 54.7 ± 15.8, respectively). The SDB between both groups did not show any differences (3.7 ± 1.5 vs 3.4 ± 1.3, respectively). The same analysis was repeated using only those patients in the 4–10 years-old range, as per the original CSHQ was validated for Owens et al., 17 but no significant differences were found either (Table 4).
CSHQ results.
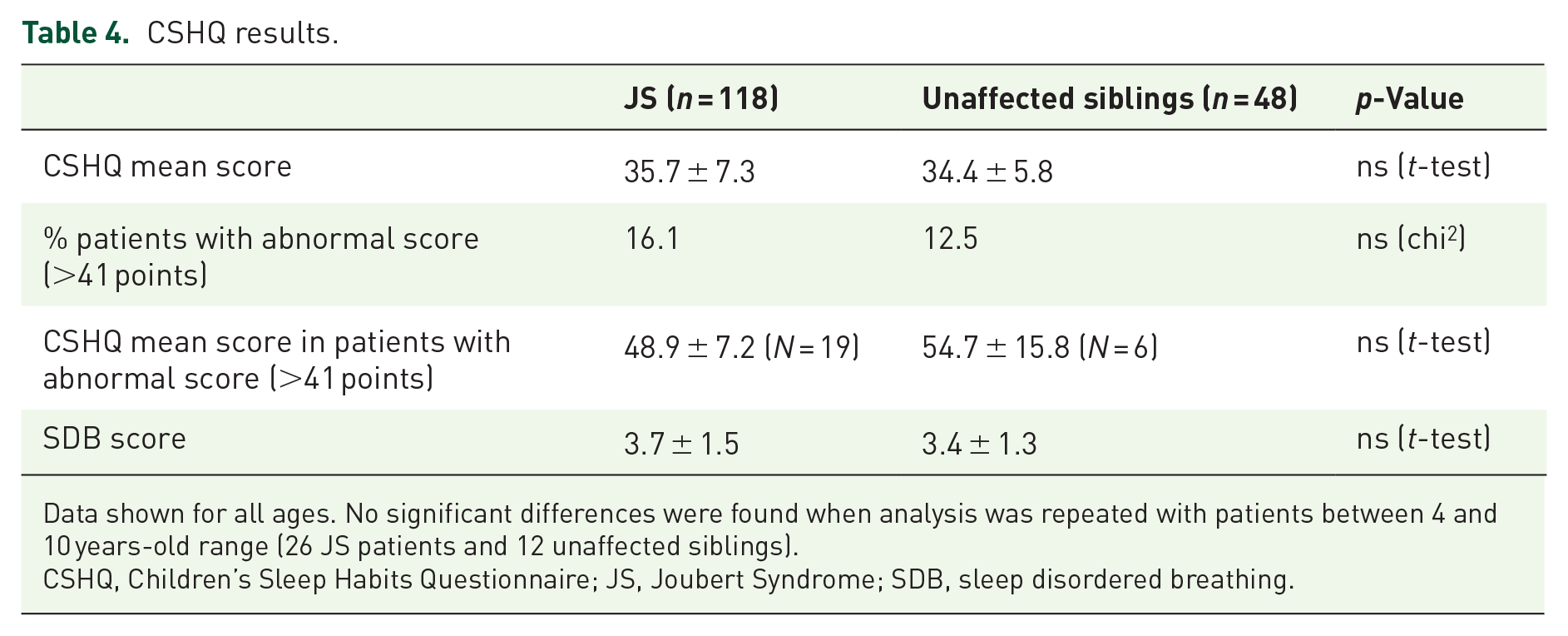
Data shown for all ages. No significant differences were found when analysis was repeated with patients between 4 and 10 years-old range (26 JS patients and 12 unaffected siblings).
CSHQ, Children’s Sleep Habits Questionnaire; JS, Joubert Syndrome; SDB, sleep disordered breathing.
There was significant difference between the mean PSQ scores among JS patients and their unaffected siblings (7.4 ± 4.1 vs 2.4 ± 2.6, respectively, p = 0.001). Among the 118 JS patients, 51 of them (43.2%) had a PSQ > 8; and among the 48 unaffected siblings, only 1 of them (2.0%) had a PSQ > 8. The mean PSQ score among those with PSQ > 8 in the JS patients’ group was 11.2 ± 2.7. The same analysis was repeated using only those patients in the 2–18 years-old range, as per the original PSQ was validated for Chervin et al., 16 and the significant difference persisted (Table 5). PSQ scores did not vary with age for both the JS and unaffected siblings’ group (data not shown).
PSQ results.

Data shown for all ages. Significant differences persisted when analysis was repeated with patients between 2 and 18 years-old range (71 JS patients and 35 unaffected siblings).
JS, Joubert Syndrome; PSG, polysomnography.
Additionally, there were no clear trends between severity of CSHQ Total Score or PSQ score, and genetic mutations described (data not shown).
Polysomnography
Eleven children with JS enrolled in the UW registry had undergone clinically indicated PSG testing prior to the time of the survey administration (Table 6). As a group, the children with JS had severe SDB characterized by a mean combined AHI of 23 ± 15, (range 5–48). Events were primarily obstructive in nature with an obstructive AHI of 18 ± 15, (range 1–45). Several also had an elevated number of central apneic events with a mean central AHI of 4 ± 4 (range 0–14). Total AHI did not correlate with age or BMI (r2 = 0.00 for both). Respiratory events were not clustered in Rapid Eye Movement (REM) sleep or while supine. The JS group had significant intermittent hypoxia with elevated desaturation indices and nadir saturations. Ventilation appeared adequate based on end-tidal carbon dioxide readings. They also had abnormal sleep architecture with increased arousal indices, decreased efficiency, and more time awake and in light sleep or wakefulness when compared to the referent data. There were no clear associations between severity of SDB and genetic mutations (data not shown). Recommended treatment for the SDB in this group was based on each individual by a treating sleep medicine provider, but included either adenotonsillectomy, continuous or variable level positive airway pressure, supplemental oxygen via nasal cannula or ongoing clinical observation alone.
PSG results compared to referent cohort; data shown in mean ± SD.

AHI, Apnea–Hypopnea Index; JS, Joubert Syndrome; PSG, polysomnography.
Nine of these children’s caregivers returned the mailed sleep survey as part of the larger study. Compared to the entire group with JS, PSQ score trended lower in this subgroup but did not reach statistical significance (7.4 ± 4.1 vs 5.4 ± 3.7, p = ns). A total of 33% of this subgroup scored above the cut-off score, compared to 43.2% in the larger group. There was no correlation between PSQ score and AHI (r2 = 0.00). CSHQ total scores (35.7 ± 7.3 vs 33.6 ± 5.2, p = ns) or SDB subdomain scores (3.7 ± 1.5 vs 3.5 ± 0.9, p = ns) did not differ between groups, nor were there any correlations between CSHQ total score or SDB subdomain score with AHI (r2 = 0.00).
Discussion
We present a systematic characterization of sleep and breathing abnormalities in a cohort of JS patients using validated caregiver reports, as well as pilot PSG data in a subgroup of children with JS. This study highlights the importance of selecting the appropriate questionnaire. The PSQ revealed marked differences between the group with JS and their unaffected siblings on the total PSQ score, suggesting the presence of more significant SDB symptomatology in the first group. Indeed, 45% of the JS group scored above the cut-off score, compared to <1% in the unaffected siblings. However, the CSHQ did not reveal any differences between groups, likely due to the wide breadth of sleep disorders covered by the CSHQ with emphasis on behavioral sleep aspects, such as bedtime resistance, sleep anxiety, onset delay, and daytime sleepiness. Moreover, the subdomain of SDB on the CSHQ was also not different between groups despite robust PSQ differences. These findings are not surprising knowing that the CSHQ questionnaire has a low sensitivity compared to the PSQ questionnaire to detect SDB. 20
PSG revealed severe SDB in a subgroup of individuals who were referred for clinical suspicion of SDB. Respiratory disturbances were primarily obstructive in nature, though central apneic events were also present, but in most cases did not constitute the majority of respiratory disturbances. This finding is vastly different than the previously believed paradigm of central respiratory control difficulties.9,10 Given objective evidence of mixed sleep apnea, authors would highly recommend performing complete PSG studies instead of limited PSG or overnight pulse oximeter to diagnose SDB in JS. Interestingly, AHI on PSG as a measure of severity of SDB did not correlate with PSQ. This is consistent with the typical pediatric population, in which parental report of SDB fails to identify obstructive sleep apnea. 21 Though numbers were overall low, there were no clear correlations between certain genetic mutations and findings on their PSG. Current literature, despite the classic description of respiratory control abnormalities in the JS population,8,10,13 shows that JS patients and their unaffected siblings had similar rates of diagnosis and treatment for SDB. This highlights the importance of using the appropriate tools to identify SDB in pediatric population.
Patients with JS exhibit a classic alternating tachypnea–apnea pattern (Figure 1), similar to periodic breathing but with a faster rate, and can happen while awake or during sleep. SDB such as central and obstructive apneas are also described, especially in patients with obesity,8,10 but they tend to attenuate with age. This was evidenced in our cohort as they had high AHI with central and obstructive events (data not shown). SDB contributes to disturbed sleep with different clinical manifestations in children. While a subset of children presents with excessive daytime sleepiness like adults, more commonly, children present with daytime cognitive and behavior problems, including difficulties with attention, aggressive behavior, and hyperactivity.22,23 Further prospective studies with PSG in symptomatic and asymptomatic patients with JS, as well as age-related trends are needed to determine the degree of impact of SDB in this population (Figure 2).

(a) Four illustrative epochs of Non-Rapid Eye Movement (NREM) sleep from a PSG performed on a 7-year-old with JS showing classic sleep disturbance pattern. Abnormal respiratory pattern with tachypnea–central apnea (red box = tachypnea; blue box = central apnea), associated with O2 desaturation and appropriate arousal (green box). This patient has chronic hyperventilation. (b) Four illustrative epochs of NREM sleep showing obstructive events in the same patient with JS. Abnormal respiratory pattern with repeated obstructive hypopneas (red box), associated with O2 desaturation. This patient has chronic hyperventilation. (c) Three illustrative epochs of NREM sleep showing mixed events in the same patient with JS. Abnormal respiratory pattern showing mixed components, with slight predominance of the obstructive component (blue box = central; red box = obstructive), and associated O2 desaturation. This patient has chronic hyperventilation.

Two illustrative epochs of wake from a PSG performed on a 2-week-old with JS showing obstructive events.
The etiology of SDB in JS is thought to be associated with the affected respiratory centers in the lower pons and medulla. The cerebellum is classically viewed as one of the primary structures involved in motor coordination and pattern generation. In addition to the motor act of breathing, the cerebellum has a possible role in central respiratory control, previously believed to be only housed in the brainstem. Breathing involves cyclic motor acts of ventilatory muscles with maintenance of neuromuscular patency of the upper airway, in response to various chemical, mechanical, and neurologic signals. In addition, the maintenance of eupneic breathing changes during times of vulnerability (e.g., sleep), and likely requires additional compensatory mechanisms beyond the rhythmogenesis associated with the brainstem’s traditional respiratory control center. Those additional fortifications may be housed in the cerebellum. The cerebellum’s role in coordination of ventilatory and airway muscles is not unexpected. 24 However, deep cerebellar nuclei, such as the fastigial nucleus, are thought to serve as integration points of autonomic tracts between the cortex and brainstem which plausibly contain respiratory signal pathways and may serve as an augmentation site when respiration is challenged. 25 Thus, children with JS may have minimal manifestations of disturbed respiration while awake and more pronounced during sleep.
Data from animal studies and human studies of healthy volunteers and those with congenital structural abnormalities provide a growing body of evidence for a complex role of the cerebellum in maintaining a eupneic state. The clinical evaluation of respiratory control classically includes ventilatory challenges to hypercapnia and hypoxia during awake and asleep states.26 –28 Unfortunately, these are often cumbersome, require patient cooperation/neurocognitive ability, and motor coordination which may be challenged in the JS population, and are limited to tertiary research settings. Because sleep is a naturally occurring state in which respiration is inherently less robust and more vulnerable than during wakefulness, the use of PSG to evaluate respiratory status has become an important clinical surrogate for other impracticable tests. Evidence of the cerebellum’s role in respiratory control is also present in those with cerebellar tumors, an unfortunate but naturally occurring population prevalent in pediatrics. A retrospective series of 36 pediatric posterior fossa tumor cases found increase in PaCO2 in 20% of patients, with 17% having documented apneic or bradypneic events documented in the first month postoperatively. 24 Interestingly, the cases with apnea had no higher incidence of brainstem involvement than the nonapnea group, further raising the possibility of cerebellar involvement in the respiratory disorder. None of these cases were evaluated with formal sleep studies.
Other contributing causes to obstructive SDB: Physiologically, individuals with JS have several risk factors for sleep-related breathing difficulties beyond the prevalence of general pediatric population. These include postulated central respiratory control abnormalities, underlying hypotonia, and airway/thoracic abnormalities, all of which predispose to poor respiratory function, which is further impaired during sleep. 29 Children with JS may likely be at even higher risk due to relative immaturity of the respiratory system, more time in total sleep and a higher percentage of time in REM sleep which is associated with more profound hypotonia.
Management of SDB has not been studied specifically in JS. Treatment varies depending on characteristics of SDB. Options include supplemental oxygen while awake and during sleep which may blunt central apneas/periodic breathing or tachypnea–apnea patterns. If hypercapnia is present, Positive Pressure Ventilation (PPV) by mask or tracheostomy may be warranted. 12 Obstructive events can be treated with Continuous Positive Airway Pressure (CPAP) or otolaryngologic procedures such as adenotonsillectomy. PPV via tracheostomy has also been described for severe cases. 12 Annual respiratory and sleep disturbances surveillance is also recommended given the risk of recurrent respiratory infections, dysphagia, restrictive lung disease due to ciliary dyskinesia, skeletal dysplasia, and hypotonia.12,30 PSG should be considered for all individuals to facilitate early identification of treatable sleep and breathing disorders. 31
Several limitations are present in our study that should be considered when interpreting the results. First, the absence of prospective data precludes the ability to compare how patients’ conditions improved or worsened with age, limiting the understanding of the natural progression of SDB in JS patients. Second, caregiver-administered questionnaires introduce the potential for recall bias, as well as performance bias, where caregivers may pay more attention to their child with JS, potentially affecting the accuracy of the reported outcomes. Third, the survey did not contemplate objective data regarding weight/BMI, which precluded further analysis to elucidate the most likely cause of the obstructive events seen in the JS patients. Additionally, the lack of access to raw PSG data as well as comparative unaffected siblings’ PSG studies restricts the depth of analysis and verification of sleep-related findings. Lastly, the genetic mutation analysis was incomplete, hindering the ability to identify specific genetic factors associated with the severity of SDB in the patient cohort. These limitations highlight the need for future research to address these gaps and provide a more comprehensive understanding of the disease and its progression. These limitations may affect the generalizability of our study.
Review of other hindbrain abnormalities and underlying pathophysiological mechanisms
Arnold–Chiari malformations
Arnold–Chiari malformations are congenital malformations characterized by herniation of the cerebellar tonsils, vermis (Chiari I), and caudal brainstem (Chiari II) through the foramen magnum, causing cerebrospinal fluid obstruction and subsequent hydrocephalus. Chiari II is typically associated with myelomeningocele.32,33 Symptoms include dysphagia, vocal cord paralysis, abnormal breathing pattern, and can be as severe as sudden infant death or lack of spontaneous breathing at birth, requiring mechanical assisted ventilation. 34
SDB is common in children with Chiari II malformation, with a prevalence as high as 81% in a retrospective study of children referred for sleep complaints (insomnia, daytime sleepiness, snoring) 34 (age 8.3 ± 0.9 years, n = 52). Central sleep apnea is the most common type of event, but hypopneas and obstructive sleep apnea has also been reported, even in an asymptomatic cohort of patients 35 (age 8.9 ± 6.2 years, n = 117). Patients with Chiari II malformation exhibit blunted hypercapnic and hypoxic ventilatory response. The exact mechanism is still unknown, but the involvement of respiratory control structures in the brainstem secondary to herniation or hydrocephalus may play a role.32,34,36
Treatment varies depending on the type of SDB present. Patients with predominantly central events and periodic breathing are treated with PPV-tracheostomy/mask or oxygen supplementation to stabilize the loop gain, while patients with more of an Obstructive Sleep Apnea (OSA) pattern will benefit from CPAP or adenotonsillectomy. In Chiari I, posterior fossa decompression and myelomeningocele improve the burden of SDB but does not resolve it completely. 37 The persistence of these events suggests a more permanent damage in respiratory control centers as the etiology of SDB.
Leigh syndrome
Leigh syndrome (LS) is a progressive neurodegenerative mitochondrial disease characterized by bilateral necrotizing lesions in the brainstem, basal ganglia, or cerebellum.38,39 Patients affected with LS are usually children under 2 years with progressive motor and neurocognitive decline, hypotonia, ataxia, ocular apraxia, dysphagia, and sleep disturbances. 40
SDB in this population are characterized by both obstructive and central events. Animal models showed blunted hypercapnic and hypoxic ventilatory responses when lesions were in the dorsal brainstem vestibular nucleus. Rhythmogenesis may also be compromised if the pre-Bötzinger complex in the ventrolateral medulla is compromised. 41 A recent study showed a significantly lower respiratory arousal index in patients with LS when compared with age-sex-AHI matched OSA patients. This suggests an inability of LS patients to arouse from sustained hypoxia triggered by SDB events, which predispose them to fatal outcomes such as sudden infant death syndrome. 42
Treatment is also targeted toward the type of respiratory disturbance during sleep. Patients with a more predominant obstructive burden are treated with CPAP, adenotonsillectomy, nasal steroids; while those with predominantly central events will benefit from supplemental oxygen or bilevel positive airway pressure to stabilize loop gain.
Pontine tegmental cap dysplasia
Pontine tegmental cap dysplasia (PTCD) is characterized by a hypoplastic ventral pons with a characteristic dorsal “cap” protruding into the fourth ventricle, thought to be caused by impaired axon migration. 43 Most patients have cognitive impairment and significant cranial nerve palsies causing corneal hypoesthesia, impaired extraocular movements, sensorineural deafness, and dysphagia.43 –45
Few studies have described SDB in patients with PTCD, predominantly Cental Sleep Apnea (CSA) that persists with time. 46 Pathological reports had shown gross abnormalities in pons and medulla, where the centers of respiratory rhythmogenesis in the ventral respiratory column and their connections are located. 47 The pons play an important role in relaying chemosensory information; thus, its hypoplasia in patients with PTCD that disrupts this transmission and leads to an increased input threshold from peripheral and central receptors to trigger a response from medulla and, subsequently, the respiratory system. 46
Treatment of SDB in patients with PTCD is similar to the aforementioned conditions, but some considerations have to be made especially due to corneal anesthesia secondary to trigeminal nerve palsy. Noninvasive positive pressure ventilation can cause desiccation and abrasions, ultimately ending in blindness. 46 Corneal neurotization surgery may aid in recovering sensation and lubrication, which would allow NIPPV. 48 Goals of therapy and shared-decision based model is critical. 49
Conclusion
In conclusion, our study underscores the common and severe nature of SDB in JS patients, with a significant predominance of obstructive events, challenging conventional perspectives on central respiratory control difficulties in this population. The observed mixed component necessitates the performance of complete PSG studies to address and prevent potential clinical manifestations in this already at-risk cohort, although the PSQ emerges as a valuable screening tool in resource-limited settings where PSG are not easily accessible. Future directions should focus on a more nuanced characterization of breathing irregularities, exploring challenges related to the central control or breathing, and conducting prospective, population-based studies within JS and JS-related disorders spectrum. Additionally, further investigations correlating SDB with genotype and radiologic findings are essential. Given the pronounced SDB burden, the study recommends routine interval surveillance for respiratory and sleep disturbances to mitigate risks associated with recurrent respiratory infections, dysphagia, restrictive lung disease, skeletal dysplasia, and hypotonia. PSG is advocated for all individuals to facilitate the early identification of treatable sleep and breathing disorders in this vulnerable population.
Supplemental Material
sj-pdf-1-tar-10.1177_17534666241308405 – Supplemental material for Sleep and breathing in children with Joubert syndrome and a review of other rare congenital hindbrain malformations
Supplemental material, sj-pdf-1-tar-10.1177_17534666241308405 for Sleep and breathing in children with Joubert syndrome and a review of other rare congenital hindbrain malformations by Jia-Der Ju-Wang, Jennifer C. Dempsey, Cristian Zhang, Daniel Doherty, Manisha Witmans, Mary Anne Tablizo and Maida Lynn Chen in Therapeutic Advances in Respiratory Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.