
Abstract
Background:
Studies have linked gut microbiota dysbiosis with sleep apnea; however, no causal relationship was found in human subjects. Finding new targets for the pathophysiology of sleep apnea might be made possible by systematically investigating the causal relationship between the human gut microbiota and sleep apnea.
Methods:
A two-sample Mendelian randomization analysis was conducted. The human gut microbiome composition data, spanning five taxonomic levels, were acquired from a genome-wide association study that included 18,340 participants from 24 cohorts. Genome-wide association study data for sleep apnea were obtained from the Sleep Disorder Knowledge Portal for primary analysis and the FinnGen consortium for meta-analysis. Sensitivity analyses were conducted to evaluate heterogeneity and pleiotropy.
Results:
Using inverse-variance weighted analysis, eight microbial taxa were initially found to be substantially linked with the apnea-hypopnea index. Only three microbial taxa remained significant associations with sleep apnea when combined with the FinnGen consortium (the class Bacilli: B = 8.21%, 95% CI = 0.93%–15.49%; p = 0.03; the order Lactobacillales: B = 7.55%, 95% CI = 0.25%–4.85%; p = 0.04; the genus RuminococcaceaeUCG009: B = −21.63%, 95% CI = −41.47% to −1.80%; p = 0.03).
Conclusions:
Sleep apnea may lead to gut dysbiosis as significant reductions in butyrate-producing bacteria and increases in lactate-producing bacteria. By integrating genomes and metabolism, the evidence that three microbiome species are causally linked to sleep apnea may offer a fresh perspective on the underlying mechanisms of the condition.
Background
Due to its high incidence, obstructive sleep apnea (OSA), a complex chronic illness, has become an important public health issue. 1 Patients with OSA frequently experience poor sleep quality, exhaustion throughout the day, memory loss, and even long-term consequences such as changes in the cardiovascular, metabolic, cognitive, and cancer-related systems. 2 Complete (obstructive apnea) or partial (hypopnea) cessation of airflow during sleep is the hallmark of OSA. This results in hypercapnia, intermittent hypoxia, and excessive negative intrathoracic pressure, which appears to be a major factor in initiating oxidative stress cascades and inflammation, ultimately leading to multi-organ morbid consequences.3,4
The gut microbiome is an essential part of the human holobiont. The gut microbiota plays a critical role in digestion and defense against invasion by pathogenic microorganisms. 5 Despite being found in the gut, the microbiota seems to play a significant role in a number of vital biological processes by generating substances like trimethylamine-N-oxide (TMAO), short-chain fatty acids (SCFAs), and primary or secondary bile acids that can be circulated and transported to remote locations.6–9 Recent studies have highlighted the link between dysbiosis and a wide range of disease states, including metabolic, neurological, behavioral, and cardiovascular diseases and cancer.6,10–12
Alterations in gut microflora richness in patients with OSA have been confirmed in cross-sectional studies.13,14 Patients with OSA have increased Firmicutes and decreased Bacteroidetes compared to the healthy population. 15 Three enterotypes, Bacteroides, Ruminococcus, and Prevotella, were identified in patients with OSA, with the latter being associated with arousal-related parameters. 16 However, other studies found altered species in patients with OSA to be different in Swedish and Hispanic populations.17,18 In addition, there is also evidence suggesting that gut microbiota dysbiosis plays a role in OSA-induced hypertension, 19 with OSA patients combined with hypertension having decreased SCFA-producing bacteria compared with controls. 20 Certain gut microbiota levels have also been linked to the development of carotid atherosclerosis and type 2 diabetes brought on by OSA.21,22 Even though gut dysbiosis and OSA have been linked in the literature, there is insufficient data to support a causal or consequential role for either condition.
Mendelian randomization (MR) is mostly implemented using single nucleotide polymorphisms (SNPs). 23 Using genetic variants in this way, MR could avoid bias from unobserved confounding factors. 24 Reverse causality is improbable because genotype creation takes place prior to the beginning of disease and is typically unaffected by disease development.
This study systematically explored the causal relationship between sleep apnea and the composition of the human gut microbiome at five taxonomic levels using a two-sample MR analysis employing previously published data from genome-wide association studies (GWAS).
Methods
Study design
The causal association between sleep apnea and the human gut microbiome composition was assessed using a two-sample MR analysis. The study overview is illustrated in Figure 1. All the analyses were performed using the R.app GUI (Version 1.79) and Review Manager (Version 5.4.1) from September to October 2023.
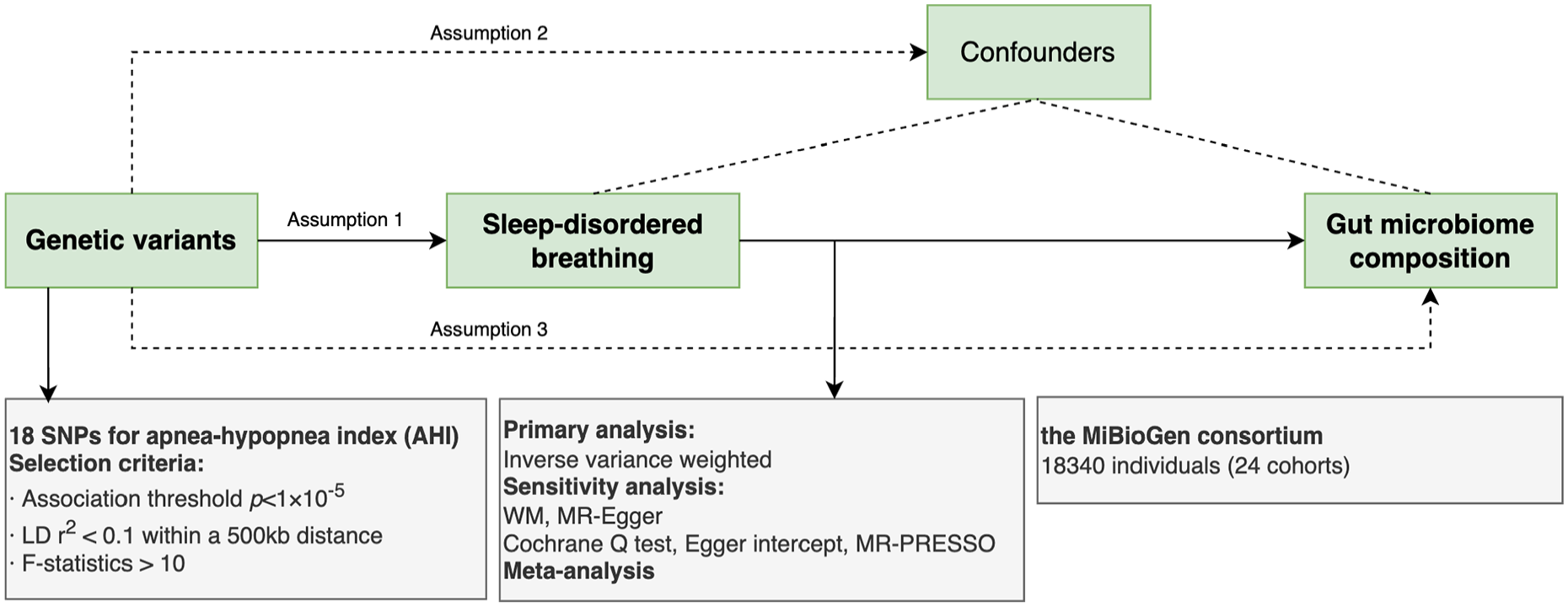
Overview of the current Mendelian randomization (MR) study.
GWAS data for sleep-disordered breathing
The primary analysis obtained the genetic information on sleep apnea from the Sleep Disorder Knowledge Portal (https://sleep.hugeamp.org/). Specifically, the genetic variants used in this study were derived from up to 5727 participants of European ancestry in five cohorts conducted by Chen et al. 25 Appropriate ethical approval and participants’ written informed consent were obtained in the original study. Sleep tests were performed by type 3 devices (n = 5101) at home or type 1 polysomnography (n = 626), and all the sleep data were scored in one Sleep Reading Center. Apneas were defined when airflow was reduced to ⩾90% of the baseline values for at least 10 s. Hypopneas were defined by a ⩾30% reduction of airflow for at least 10 s accompanied by at least a 4% drop in oxygen saturation. The apnea-hypopnea index (AHI), which is based on the number of apnea and hypopnea events per hour of sleep, was used to measure the severity of sleep apnea. Individuals who were under treatment of continuous positive airway pressure (CPAP), overnight oxygen, or oral devices for sleep apnea were excluded. In total, OSA was diagnosed in 2164 participants.
To validate our results by conducting replication analysis and meta-analysis, the sleep apnea data (ICD-10 code G47.3) were used from Freeze 7 of the FinnGen consortium (27,207 sleep apnea cases and 280,720 healthy controls), which is publicly available on the website: https://www.finngen.fi.
GWAS data for human gut microbiome
The GWAS statistics for human gut microbiome composition were obtained from the MiBioGen consortium at https://www.mibiogen.org, comprising 18,340 individuals from 24 cohorts (nearly 86.3% of Europeans and multiethnic groups). The ratio of males and females was almost 1:1. Appropriate ethical approval and participants’ written informed consent were obtained in the original study. The microbiome structure at multiple taxonomic levels (from phylum to genus) was investigated to explore gut microbial diversity. The original GWAS study included more details about quality control and demographic data. 26
Instruments selection
First, previous literature27–29 was referenced, the association threshold was relaxed to p < 1 × 10−5, and pairwise linkage disequilibrium (LD) r2 < 0.1 within a 500 kb distance was conducted to obtain top independent SNPs, due to the limited number of SNPs reaching p < 5 × 10−8. The total F-statistics reached 22.5 (>10), indicating relatively strong instruments. Second, the exposure SNPs were extracted from the outcome data, and those associated with the outcome (p < 1 × 10−5) were excluded. Proxies search was not used for SNPs absent in the outcome. Third, harmonization was conducted to align the alleles of exposure and outcome SNPs, and SNPs with intermediate effect allele frequencies (EAF) > 0.42 or with incompatible alleles were discarded.
Causal association analyses
The random-effect inverse variance weighted (IVW) method was used as the primary analysis to identify significant causal associations between sleep apnea and gut microbiome composition with p < 0.05. Due to its pleiotropic bias susceptibility and assumption that all genetic variants are legitimate, IVW is thought to be the most effective method for MR estimation. 30
For the identified significant estimates (IVW p < 0.05), sensitivity analyses were conducted using weighted median (WM) and MR-Egger to evaluate any bias of the MR assumptions. The presence of heterogeneity was conducted with the Cochran Q test, with Cochran-Q derived p < 0.05 and I2 > 25% recognized as an existing heterogeneity. 31 MR Pleiotropy RESidual Sum and Outlier findings were used to eliminate significant outliers (MR-PRESSO). The Egger intercepts were used to assess horizontal pleiotropy. 32
Consequently, the following criteria were met to estimate the possible relationship between sleep apnea and the gut microbiota: (1) consistent directions among the three MR methods; (2) no heterogeneity or pleiotropy was detected; and (3) no significant outliers were identified in MR-PRESSO.
Meta-analysis
To validate the robustness of candidate causal association, IVW analysis was replicated using independent sleep apnea GWAS data from the FinnGen consortium, and a random-effect meta-analysis was conducted to determine the final results.
Confounding analysis
SNPs were scanned with the Phenoscanner V2 website (http://www.phenoscanner.medschl.cam.ac.uk/) to explore whether the SNPs representing sleep apnea were associated with several common risk factors that might bias the MR estimates, including smoking, 33 obesity, 34 diabetes, 35 and hypertension. 36 Once the SNPs were associated with these potential confounders at the threshold of p < 1 × 10−5, the IVW estimation was replicated with the SNP discarded.
Results
Study population
Supplemental Tables 1 and 2 present the features of the studies from which GWAS data on sleep apnea and the composition of the human gut microbiome were derived. Overall, a total of 5727 participants of European descent from five cohorts were included, of which 2164 (37.8%) were diagnosed with untreated OSA, with an average AHI from 5.59 to 12.73 events/h. The average age was 65.78 ± 14.57 years, with 31.9% being females. The average BMI was 28.24 ± 5.33 kg/m2.
Primary analysis
The number of SNPs for AHI and sleep apnea was 18 and 9, respectively. The harmonized data are presented in Supplemental Table 3.
Eight microbial taxa were preliminarily identified as significantly associated with AHI by IVW analyses (Figures 2 and 3), including the class Bacilli, the orders Bifidobacteriales and Lactobacillales, the family Bifidobacteriaceae, and the genera Bifidobacterium, Ruminiclostridium6, Rumino-coccaceaeUCG009, and unknown (ID 826). No significant association was found at the phylum level. Specifically, AHI was found to increase the abundance of the class Bacilli (B = 9.07%, 95% CI = 0.66%–17.51%), and its order Lactobacillales (B = 8.57%, 95% CI = 0.77%–16.37%); the order Bifidobacteriales (B = 9.08%, 95% CI = 0.66%–17.51%), its family Bifidobacteriaceae (B = 9.08%, 95% CI = 0.66%_17.51%), and its genus Bifidobacterium (B = 9.82%, 95% CI = 1.35%–18.28%). Furthermore, AHI was also found to increase the abundance of the genus Ruminiclostridium6 (B = 9.13%, 95% CI = 0.29%–17.96%), but decrease the genus RuminococcaceaeUCG009 (B = −15.43%, 95% CI = −27.34% to −3.43%) and unknown (ID 826) (B = −9.14%, 95% CI = −18.01% to −0.28%). Except for two microbial taxa, MR estimates obtained from WM and MR-Egger regression demonstrated consistent direction and magnitude, demonstrating the general robustness of the causality (Table 1). No instruments were excluded by the results of MR-PRESSO.

Forest plot of the causal effect of AHI on human gut microbiome composition derived from inverse variance weighted (IVW).

Scatterplot of the significant Mendelian randomization (MR) association (p < 0.05) between AHI and human gut microbiome composition.
Primary and sensitivity analyses for the causal association between AHI and gut microbiome composition.

AHI: apnea-hypopnea index; IVW: inverse-variance weighted; WM: weighted median; B: beta; CI: confidence interval.
Meta-analysis
Replication analysis was done utilizing sleep apnea GWAS data from the FinnGen collaboration to confirm the meta-analysis results. As shown in Figure 4, the genetic predisposition of sleep apnea predicted increased class Bacilli abundance (B = 8.21%, 95% CI = 0.93%–15.49%; p = 0.03), increased order Lactobacillales abundance (B = 7.55%, 95% CI = 0.25%–14.85%; p = 0.04), and decreased genus RuminococcaceaeUCG009 abundance (B = −21.63%, 95% CI = −41.47% to −1.80; p = 0.03). In the meta-analysis, the correlation between sleep-disordered breathing and the remaining microbial taxa did not become statistically significant.

Meta-analysis of the causal associations between sleep apnea and human gut microbiome composition.
Confounding analysis
Although sensitivity analysis revealed no evidence of bias invalidating the MR estimates, we further investigated the second traits (smoking, obesity, diabetes, and hypertension) of the AHI-associated SNPs. The SNPs associated with AHI were not associated with any of the above-mentioned confounders on the Phenoscanner.
Discussion
This study looked for possible causal relationships between gut microbial taxa and sleep disorders by using meta-analysis and two-sample MR analysis. The findings revealed a causative relationship between higher AHI and genetic predisposition for sleep apnea, as well as a rise in the class Bacilli and order Lactobacillales and a drop in the genus RuminococcaceaeUCG009.
The symptoms of sleep apnea include intermittent hypoxia and repeated airflow cessation during sleep. These conditions can create an anaerobic environment and inadequate oxygen delivery to the intestinal mucosa, which can cause intestinal mucosal ischemia-reperfusion injury and even systemic low-grade inflammation.13,37 Anaerobic Gram-negative bacteria, which disrupt the structure of the intestinal microbial community and alter the composition of gut bacteria, also benefit ecologically from the change in PaO2. 38 Sleep fragmentation, another key feature of OSA caused by recurrent episodes of apnea and hypopnea, might also play a role in microbiome change in OSA. 16 Numerous sleep apnea-mimicking animal paradigms and human observational investigations have revealed conditions such as reduced microbial diversity, dysbiosis, and changes in inflammation-related strains.3,19,39–43 However, a causal relationship between OSA and systemic diseases intermediated by gut dysbiosis, such as hypertension and cognitive impairment, comes from animal studies, whereas a direct cause-and-effect relationship has not been previously established in humans.
This study found that sleep apnea increased the abundance of the order Lactobacillales. Lactobacillales, a member of the Bacilli class, are primarily metabolic producers of lactic acid and are typically found in milk products and decaying plants. Low circulating levels of lactic acid are found in healthy individuals. 44 However, previous animal paradigms have confirmed an increase in the level of lactate in a chronic intermittent hypoxia animal model, and in the relative abundance of lactate-producing genera Lactococcus led by a high-fat diet.19,45. Elevated blood pressure and a higher AHI are linked to serum lactate levels.37,46 Since the plasma lactate acid level is significantly associated with inflammatory mediators (e.g., interleukin-1β, tumor necrosis factor-α), it has been considered a reflection of intestinal mucosal permeability and damage degree in OSA patients. Thus, our findings may indicate that primarily anaerobic metabolism in sleep apnea may lead to a subclinically disturbed intestinal barrier and its hyperpermeability, most likely through the action of Lactobacillales and an increase in lactate. Furthermore, the hypertension brought on by OSA may also be attributed to the order of Lactobacillales. 19
This study suggested that the genetic liability of sleep apnea could lead to a decrease in the genus RuminococcaceaeUCG009. One of the most predominant families within the order Clostridiales in the mammalian gut environment is the Ruminococcaceae, which is capable of breaking down the cellulose and hemicellulose components of plant matter as well as producing large amounts of SCFA, which lowers inflammation by preserving the integrity of the gut barrier.47–49 Produced by bacterial fermentation of dietary fibers in the gut, butyrate makes up over 95% of SCFAs and has been demonstrated to be crucial for iron absorption, cardioprotection, blood pressure regulation, obesity reduction, and improved insulin sensitivity.9,50–52 Wang et al. 43 have observed genera involved in the production of SCFAs to be significantly decreased in sleep fragmentation patients and animal models, who also confirmed gut microbiota regulate cognitive functions through SCFAs. As a prominent producer of butyrate, Ruminococcaceae was decreased in relative abundance in OSA-induced hypertension rats, 19 and in patients with liver diseases.47,53,54 Therefore, the genus RuminococcaceaeUCG009 might also play a part in OSA-induced hypertension and cognitive impairment.
By reducing the genus RuminococcaceaeUCG009 and raising the lactate-producing metabolism due to the abundance of the class Bacilli and the order Lactobacillales, sleep apnea may result in a downgraded butyrate metabolism pathway, which may further exacerbate hypertension (Figure 5). These findings were consistent with previous OSA-induced hypertension-mimicking animal paradigms.19,42 However, previous studies have also confirmed other butyrate- and lactate-producing microbiome taxa, such as the genus Eubacterium, Coprococcus, Pseudobutyrivibrio, Streptococcus, and Turicibacter to be altered in OSA,19,55 which were not found statistically altered in this study. In addition, several bacterial species create other SCFAs like propionate and acetate, which help preserve gut health and equilibrium. The current study did not examine other dysbiosis-related metrics, such as the Shannon index and the F/B ratio (Firmicutes/Bacteroidetes) of the microbial communities in OSA patients.42,55 Therefore, the preliminary findings of this study need to be strengthened by future studies.
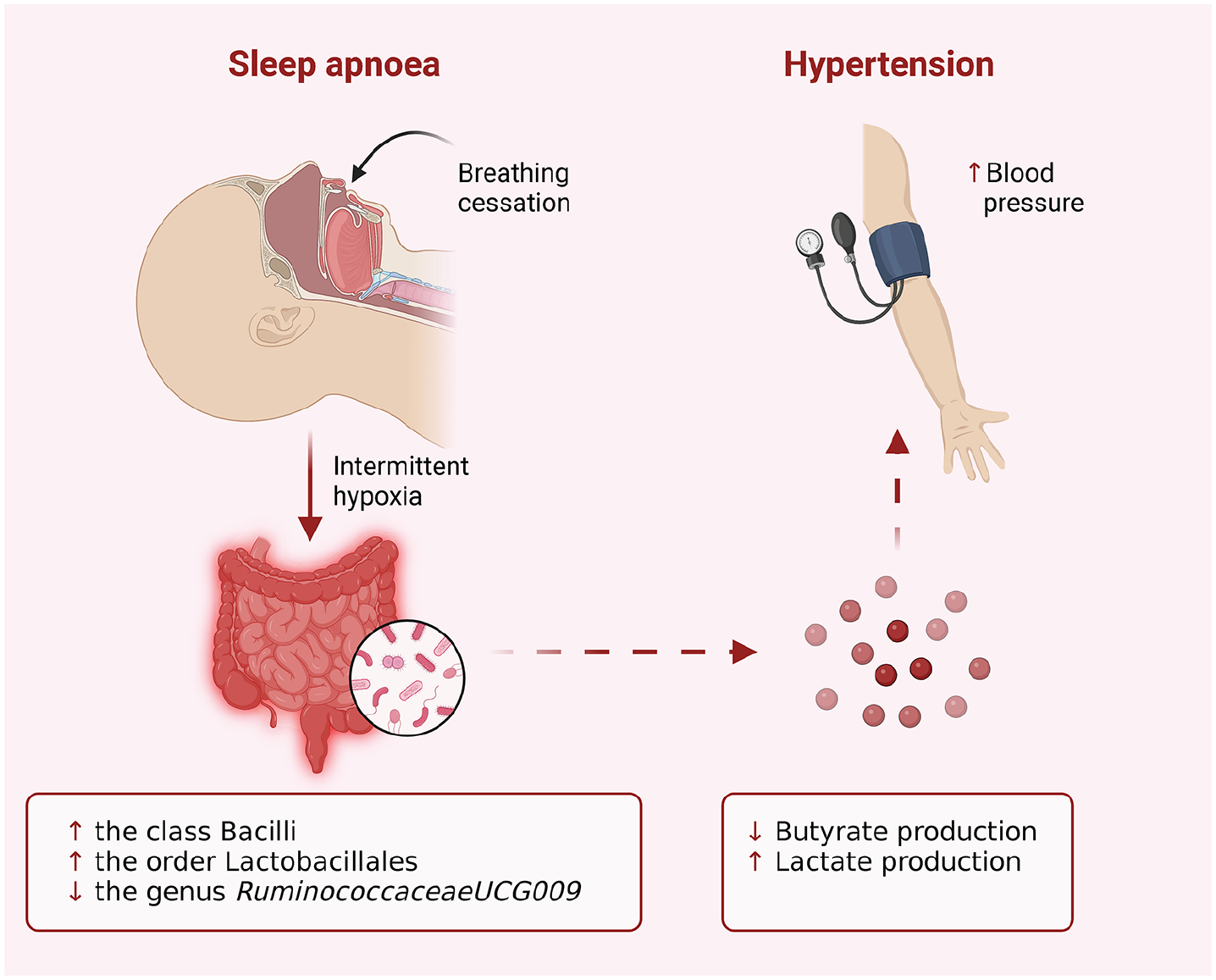
The postulated pathophysiology of OSA-induced hypertension from gut microbiome dysbiosis.
The current study has a number of advantages. This study contributed to a thorough causality analysis by examining a broad range of microbiome taxonomic levels, from phylum to genus. Second, this study is largely free from reverse causation and residual confounding due to the design of the MR. Moreover, a meta-analysis was conducted to bolster the causal relationship between sleep apnea and gut microbiota composition.
However, there are a few limitations to be aware of with this study. First, the subjects’ regular diet and antibiotic exposure were not considered, which could have an impact on the microbiota’s makeup. 56 The conditions of systematic diseases of the participants, as well as other sleep-related parameters, such as comorbidities and oxygen desaturation index (ODI), were not considered, either. Second, the majority of the participants in the study were European. As the incidence and predisposing factors of sleep apnea varied across ethnicities, 57 the causality confirmed in this study might be limited. Third, the study’s p-threshold was relaxed, a regularly used technique, due to the small number of SNPs achieving genome-wide significance. The total F-statistics reached 22.5, indicating relatively strong instruments. Finally, despite using SNPs as proxies to reduce potential bias, the underlying mechanisms of the causal association documented in the study require further validation in well-designed studies, such as experimental modes of treatment.
Conclusions
Due to notable decreases in butyrate-producing bacteria and increases in lactate-producing bacteria, sleep apnea may cause gut dysbiosis. By fusing genomes and metabolism, the finding that three microbiome species are causally linked to sleep apnea may offer a fresh perspective on the underlying mechanisms of the condition. It might be beneficial for clinicians to pay more attention to the gut microbiome of sleep apnea patients.
Supplemental Material
sj-xlsx-1-smo-10.1177_20503121241248044 – Supplemental material for Assessing the causal association between sleep apnea and the human gut microbiome composition: A two-sample Mendelian randomization study
Supplemental material, sj-xlsx-1-smo-10.1177_20503121241248044 for Assessing the causal association between sleep apnea and the human gut microbiome composition: A two-sample Mendelian randomization study by Min Yu, Xuehui Chen, Xin Huang and Xuemei Gao in SAGE Open Medicine
Footnotes
Acknowledgements
We would like to thank all the participants and investigators of Chen’s study, the FinnGen consortium, and the MiBioGen consortium.
Authors’ contributions
Yu M designed the work, analyzed and interpreted the data, and drafted and substantively revised the manuscript. Chen XH and Huang X interpreted the data. GXM substantively revised the manuscript. All authors read and approved the final manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplemental information file.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Program for Multidisciplinary Cooperative Treatment on Major Diseases (PKUSSNMP-201902).
Ethics approval
Ethical approval was not sought for the present study because appropriate ethical approval was obtained in the original study.
Informed consent
Informed consent was not sought for the present study because participants’ written informed consent was obtained in the original study.
Consent for publication
Not applicable.
Trial registration
Not applicable.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.