
Abstract
Joubert syndrome (JS) is a recessive disorder that is characterized by midbrain-hindbrain malformation and shows the “molar tooth sign” on magnetic resonance imaging. Mutations in 40 genes, including Abelson helper integration site 1 (AHI1), inositol polyphosphate-5-phosphatase (INPP5E), coiled-coil and c2 domain-containing protein 2A (CC2D2A), and ARL2-like protein 1 (ARL13B), can cause JS. Classic JS is a part of a group of diseases associated with JS, and its manifestations include various neurological signs such as skeletal abnormalities, ocular coloboma, renal disease, and hepatic fibrosis. Here, we present a proband with the molar tooth sign, ataxia, and developmental and psychomotor delays in a Dagestan family from Russia. Molecular genetic testing revealed two novel heterozygous variants, c.2924G>A (p.Arg975His) in exon 28 and c.1241C>G (p.Pro414Arg) in exon 12 of the transmembrane protein 67 (TMEM67) gene. These TMEM67 gene variants significantly affected the development of JS type 6. This case highlights the importance of whole exome sequencing for a proper clinical diagnosis of children with complex motor and psycho-language delays. This case also expands the clinical phenotype and genotype of TMEM67-associated diseases.
Keywords
Introduction
Joubert syndrome (JS, OMIM PS213300) is a recessive disorder that is clinically and genetically heterogeneous. The disorder was first described in 1969 in a patient with abnormal tremor, hypotonia, and agenesis of the cerebellar vermis. 1 This neurodevelopmental ciliopathy is accompanied by dysregulation of breathing, hypotonia, ataxia, abnormal eye movements, and cognitive impairment. 2 The main radiological feature of the disease is a “molar tooth sign” observable by magnetic resonance imaging (MRI).
The variety of symptoms of the syndrome can be partly explained by the genetic basis of the disease. Mutations in more than 35 genes, including Abelson helper integration site 1 (AHI1), inositol polyphosphate-5-phosphatase (INPP5E), coiled-coil and c2 domain-containing protein 2A (CC2D2A), and ARL2-like protein 1 (ARL13B), 3 are responsible for autosomal recessive JS, and one gene causes X-linked JS. 4 These genes encode primary cilia proteins, and mutations in each gene cause a different type of JS. 5 Classic JS belongs to a group of Joubert syndrome-related disorders (JSRDs), manifestation of which is characterized by various neurological signs including skeletal abnormalities, ocular coloboma, retinal dystrophy, renal disease, and hepatic fibrosis. 6 Mutations in the following genes are most commonly associated with the development of pathology: INPP5E (JBTS1, OMIM 613037), TMEM216 (JBTS2, OMIM 613277), AHI1 (JBTS3, OMIM 608894), NPHP1 (JBTS4, OMIM 607100), CEP290 (JBTS5, OMIM 610142), transmembrane protein 67 (TMEM67) (JBTS6, OMIM 609884), RPGRIP1L (JBTS7, OMIM 610937), ARL13B (JBTS8, OMIM 608922), CC2D2A (JBTS9, OMIM 612013), and OFD1 (JBTS10, OMIM 300170). The estimated JSRD incidence ranges from 1:80,000 to 1:100,000. 7
In this report, we present a Caucasian proband with the molar tooth sign recognizable on MRI, ataxia, and speech and developmental delays. Molecular analysis of the parents revealed two novel heterozygous variants, c.2924G>A (p.Arg975His) in exon 28 and c.1241C>G (p.Pro414Arg) in exon 12 of the TMEM67 gene. According to the literature, mutations in CPLANE1, CC2D2A, AHI1, CEP290, and TMEM67 occur in 6% to 9% of JS cases. 6 In the homozygous and compound heterozygous state, TMEM67 mutations can lead to the development of several diseases, including JS type 6 (OMIM 609884). This case expands the clinical phenotype and genotype of TMEM67-associated diseases.
Case report
The proband was a 5-year-old Kumyk girl from the Republic of Dagestan, Russian Federation. She was born from the second pregnancy (first pregnancy – miscarriage at 7–8 weeks) of healthy parents who denied consanguinity. The family history was unremarkable. An acute respiratory viral infection with elevated body temperature occurred during the early stages of the pregnancy. The proband was born at 40 weeks via an urgent vaginal delivery. She had a birth weight of 3500 g, head circumference of 35 cm, height of 51 cm, and Apgar score of 7/8. A developmental delay had been noted since birth. At the age of 3 months, she would not fix her eyes on her toys, had no facial expressions, and lacked emotions. The proband was able to sit independently and walk with support from 17 months of age. A physical evaluation at the age of 24 months showed motor and psycho-language delays and tremor of the upper limbs. By 36 months, the proband could walk independently for a short distance. An MRI examination performed at 27 months showed the molar tooth sign (Figure 1).

(a) The phenotype of a 5-year-old girl with Joubert syndrome and divergent strabismus and (b) Magnetic resonance imaging of the cerebellum and brain stem revealed the “molar tooth sign” (red arrow).
According to the clinical examination conducted at 4 years of age, the proband could stand and walk independently and speak several words. She had a height of 103 cm (50th–75th centile), weight of 17 kg (50th–75th centile), head circumference of 49 cm (10th–25th centile), and chest circumference of 55 cm (50th–75th centile). Low-set large outer ears were noted as a minor developmental anomality. Regarding the skull shape, the patient had distinctive parietal tubercles and a sloping occiput. She was determined to have intermittent divergent strabismus of the left eye, chorioretinitis, and retinal angiopathy. Electroencephalography data were normal, and foci of epileptic activity were absent. Ultrasound examination of the abdominal cavity, liver, kidney, and heart did not show any significant abnormalities. Electromyoneurography revealed impaired suprasegmental control of motor activity in the lower limb muscles and an increased phasic component of gastrocnemius muscle tone on both sides. Amplitudes of proximal and distal motor responses were normal. After consultation with a geneticist, she was referred for whole exome sequencing (WES), which revealed two heterozygous variants in TMEM67 that have not been identified previously: NM_153704.6, c.1241C>G (GRCh38.p13 – Chr8:93785331) and c.2924G>A (GRCh38.p13 – Chr8:93816388) (Figure 2).

(a) Pedigree of the 5-year-old female proband’s family with two heterozygous variants in TMEM67: c.2924G>A (p.Arg975His) and c.1241C>G (p.Pro414Arg). (b) Sequencing chromatogram of the proband’s father and sister showing the heterozygous missense mutation c.2924G>A (p.Arg975His) in TMEM67. (c) Sequencing chromatogram of the proband’s mother showing the heterozygous missense mutation c.1241C>G (p.Pro414Arg) in TMEM67. (b, c) Alignment of next-generation sequencing reads against a reference genome (GRCh37.p13) was performed for the proband. The proband was heterozygous for both the c.2924G>A and c.1241C>G pathogenic variants. Circle – female, square – male, open symbols – unaffected, filled symbols – affected, arrow – proband. The bases in the grey frame are mutational sites.
According to the clinical evaluation conducted at the age of 5, the patient had a height of 105 cm (25th–50th centile), weight of 18 kg (50th centile), head circumference of 51 cm (50th–75th centile), and chest circumference of 55 cm (50th–75th centile). Other evaluations performed on the 5-year-old proband included abdominal ultrasonography and ophthalmologic, neurological, and neuropsychological evaluations. The intermittent divergent strabismus of the left eye, chorioretinitis, and retinal angiopathy showed no dynamic changes. The neurological examination revealed dynamic ataxia. She walked independently and spread her legs wide during walking. The muscle tone of the proband was evaluated using the original Medical Research Council scale. She had a slight reduction in muscle tone in her upper and lower limbs. Her muscle strength was rated at 4.5 points. The proband had symmetrical reduction of tendon and periosteal reflexes, but pathological reflexes were not detected. Underdevelopment of pelvic function was noted. Assessment of psycho-speech development indicated a lack of speech. The child selectively followed simple instructions. The lack of speech was compensated for by facial expressions and gestures. She was not interested in playing together or communicating with peers. When examining and addressing the child, a specific reaction was noted: stereotypy in the muscles of the face in the form of “grimacing” (snarling and closing her eyes) accompanied by clenching and unclenching the fists.
The clinical severity of the proband was quantified using two clinical scales: the Luria–Nebraska Neuropsychological Battery adapted by Glozman J.M. 8 and the Gross Motor Function Classification Scale. Lurian analysis helps to differentiate learning and behavior difficulties in accordance with the individual characteristics of brain structure functioning. The proband’s attention score was low (1–2 points) compared with that of individuals with normal function (5–7 points). The patient had unstable attention, a low psychomotor score (3 points compared with a normal level of 12–15 points), fixed attention, and slow switchover between objects. Direct mechanical visual memory was reduced to three objects (compared with a normal score of seven to eight objects). The coefficient of mediated memorization was 37%, which is lower than the normal rate (73%). Speech was absent. The patient’s drawings were sloppy and chaotic. Her dynamic and motivational-personal aspects of reasoning were not affected. The proband could partially classify objects, recognize some animals, and make their sounds.
Whole exome sequencing
WES was performed to identify the genetic cause of the patient’s disease, and Sanger sequencing was performed on her mother, father, and youngest sister, who had no manifestations of the disease. Genomic DNA from peripheral blood samples was isolated using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). DNA libraries were prepared using a QIAseq FX DNA Library Combinatorial Dual-Index Kit (Qiagen). The samples were enriched using a SureSelect XT2 kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced using a HiSeq 2500 sequencer (Illumina, San Diego, CA, USA), generating 2 × 100 base pair reads (see Supplementary materials, Supplementary methods). After sequencing, 3′-nucleotides with a read quality less than 10 were trimmed using Cutadapt. 9 Illumina raw reads were aligned to reference genome hg19 (GRCh37.p13) using the Burrows–Wheeler Aligner Maximum Entropy method. The aligned file was used for variant calling with the Genome Analysis Toolkit according to Genome Analysis Toolkit best practices. 10 FastQС was used for data quality control. 11
We identified two novel (p.Pro414Arg and p.Arg975His) heterozygous substitutions within the TMEM67 protein, and these variants had not been previously identified in the ExAC and 1000 Genomes browsers. The heterozygous variant c.2924G>A was previously found in one Genotek Ltd. patient from the Russian population among our 3000 in-house exomes. The combination of the two novel compound heterozygous variants was found only in the proband. These variants were inherited by the patient from her parents, which was confirmed by Sanger sequencing (Figure 2a). The first TMEM67 variant (c.2924G>A, GRCh38.p13 – Chr8:93816388, NM_153704.6) was classified as having “uncertain significance” according to the variant interpretation guidelines of the American College of Medical Genetics (PM2, PP3). 12 The variant was classified as “pathogenic” according to the predictive programs SIFT, PolyPhen2, and Mutation Taster (PolyPhen2 HumDiv converted rank score 0.91; PolyPhen2 Hum Var converted rank score 0.81; SIFT converted rank score 0.68; Mutation Taster converted rank score 0.81). The father and sister of the proband were healthy and carried this variant in the TMEM67 gene (Figure 2b). The second TMEM67 variant (c.1241C>G, GRCh38.p13 – Chr8:93785331) was classified as having “uncertain significance” according to the variant interpretation guidelines of the American College of Medical Genetics (PM2, PP3). The variant was classified as “pathogenic” according to the predictive programs SIFT, PolyPhen2, and Mutation Taster (PolyPhen2 HumDiv converted rank score 0.91; PolyPhen2 Hum Var converted rank score 0.97; SIFT converted rank score 0.91; Mutation Taster converted rank score 0.81). The mother of the proband was healthy and carried this variant in the TMEM67 gene (Figure 2c). The list of variant effect predictor results from the in silico analysis of the pathogenicity of the identified mutations is presented in Supplementary Table 1.
This case report conforms to the CARE guidelines. 13
Discussion
We present a case report of a proband with compound heterozygous mutations in the TMEM67 gene identified in a WES study. According to the literature, JS, similar to other ciliopathies, is highly heterogeneous in its molecular basis and clinical manifestations. Dysfunctions of several organ systems, such as renal disease and hepatic fibrosis, may develop with a delay. 14 TMEM67 is one of the five main genes (CPLANE1, CC2D2A, AHI1, CEP290, and TMEM67) in which mutations lead to the development of JS (6%–9% of cases). In most cases, pathogenic variants in TMEM67 are associated with JSRD with liver involvement and/or coloboma. 15 The severity of symptoms varies from mild to severe, such as hepatic fibrosis.
The TMEM67 gene plays a role in ciliogenesis 16 and encodes the transmembrane protein TMEM67/meckelin. Primary cilia regulate chemo-, mechano-, and photoreception 17 signaling pathways during embryogenesis and postnatal tissue homeostasis. 18 Primary cilia have complex structures, and their components are responsible for their formation and function. Mutations in genes encoding proteins that are structural or functional components of cilia lead to the development of ciliopathies of various origins. 19 TMEM67 is one of the proteins in the transition zone that forms a diffusion barrier at the base of the cilium that restricts entrance and exit of both membrane and soluble proteins.20,21 TMEM67 consists of 995 amino acids and includes an N-terminal cysteine-rich domain with a highly conserved cysteine-rich repeat domain, seven transmembrane domains, and a predicted β sheet-rich region. 22
Two novel compound heterozygous TMEM67 gene variants, p.Arg975His in exon 28 and p.Pro414Arg in exon 12, were identified. The heterozygous variant p.Arg975His was also found in a 9-year-old girl from the same region. The proband was a Dargin girl from the Republic of Dagestan, Russian Federation. WES was performed because of suspected growth retardation. The girl did not have a developmental delay, and her MRI findings were normal. Thus, this child was a heterozygous carrier of the identified variant, c.2924G>A (p.Arg975His), in the TMEM67 gene. Usually, variants and their combinations are unique for each family. Considering the ethnic characteristics of the children from the described region, the detected variant, c.2924G>A (p.Arg975His), in exon 28 may have a similar origin and be a founder mutation. However, the lack of sufficient molecular data does not allow determination of the age of the mutation and the time of divergence of these peoples.
The affected parts of the gene are highly conserved across species, confirming the pathogenicity of these variants (Figure 3a). The proline at position 414 of the protein is located in the extracellular β sheet-rich domain, which connects the N-terminal cysteine-rich domain and transmembrane domain of the protein and could mediate signal transduction. Mutations in this domain can destabilize the protein structure and lead to conformational changes. 23 The arginine at position 975 of the protein is located in the transmembrane domain of meckelin (Figure 3b). 23
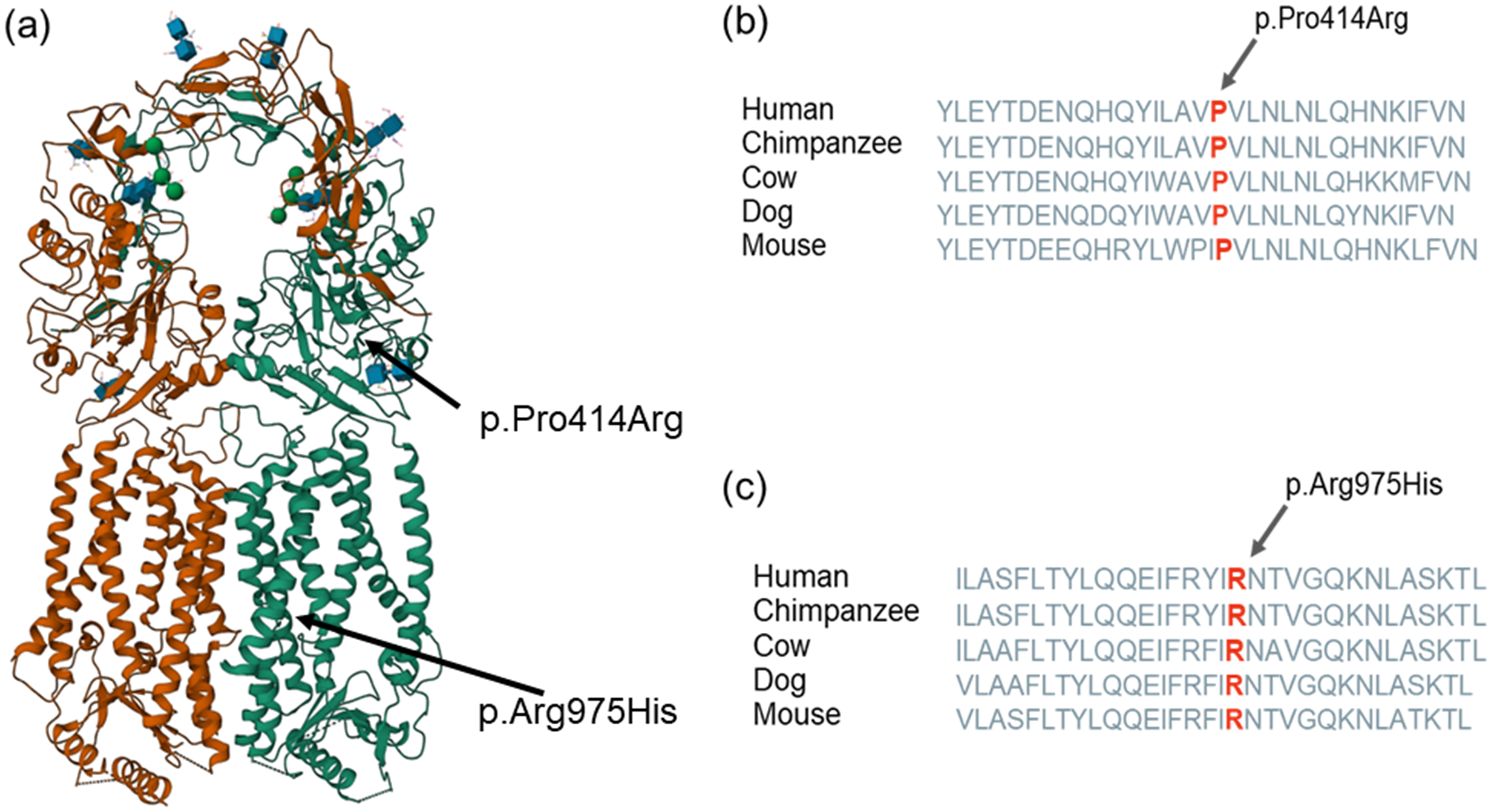
(a) Structural mapping of the two novel pathogenic mutations onto the structure of meckelin. The protein is a homodimer, and protomers A and B are shown in green and orange, respectively. Carbohydrates are shown with blue cubes and green beads (the structure is from the Protein Data Bank, https://www.rcsb.org/3d-view/7FH1/1) and (b, c) Conservation of Pro414 and Arg975 in different species of mammals. The arrows and red letters indicate the affected site.
Mutations in the TMEM67 gene can lead to the development of several diseases including RHYNS syndrome (OMIM 602152), COACH syndrome 1 (OMIM 216360), JS type 6 (OMIM 610688), and Meckel syndrome 3 (OMIM 607361). Based on the clinical examination and genetic analysis, the proband was diagnosed with JS type 6. This subtype is a genetically distinct form of JS and is characterized by hypotonia, mental retardation, ataxia, oculomotor apraxia, retinal, renal, or hepatic degeneration, and vermis hypoplasia or aplasia.24,25 In contrast, our patient had poor language ability and developmental delay but no kidney or liver damage as assessed by an ultrasound examination. Further continuous monitoring of the proband is needed, but the disease exhibits a less severe form because the identified variants in exons 12 and 28 are not the most common gene hotspots. Among the 28 exons that make up the TMEM67 gene, up to 60% of mutations occur in exons 2, 6, 8, 11, 13, 15, 18, and 24. All previously identified variants in the TMEM gene are presented in Supplementary Table 2. According to the literature, several lethal phenotypes tend to be observed in probands with combinations of nonsense/missense variants. Additionally, the prevalence of missense mutations in the gene depends on the disease (JS or Meckel syndrome). Thus, in Meckel syndrome, most missense mutations are located in exons 8 to 15, whereas in JS, only one-third are found in these exons. To date, only mutation of the stop codon of exon 12 has been described. 26 Two stop codon and missense mutations were found in exon 28. 27 Thus, these novel compound heterozygous variants have never been documented in the literature.
This case highlights the importance of exome sequencing for proper clinical diagnosis of children with psycho-motor delay and JS associated with mutations in the TMEM67 gene. Usually, most TMEM67 gene variant combinations are unique in the development of ciliopathy. 26 Considering the ethnicity of the proband’s family and the characteristics of the culture and planning for childbearing, which is typical for this region, early diagnostics will allow for pregnancy planning to take into account the high genetic risk associated with the diagnosis.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605231206294 - Supplemental material for A case of Joubert syndrome caused by novel compound heterozygous variants in the TMEM67 gene
Supplemental material, sj-pdf-1-imr-10.1177_03000605231206294 for A case of Joubert syndrome caused by novel compound heterozygous variants in the TMEM67 gene by Anastasiya Aleksandrovna Kozina, Guria Kurbanovna Kanaeva, Natalia Vladimirovna Baryshnikova, Anna Yurievna Ilinskaya, Anna Alexandrovna Kim, Anastasia Vladimirovna Erofeeva, Nadezhda Andreevna Pogodina, Jamilya Payzutdinova Gadzhiyeva, Ekaterina Ivanovna Surkova and Valery Vladimirovich Ilinsky in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_03000605231206294 - Supplemental material for A case of Joubert syndrome caused by novel compound heterozygous variants in the TMEM67 gene
Supplemental material, sj-pdf-2-imr-10.1177_03000605231206294 for A case of Joubert syndrome caused by novel compound heterozygous variants in the TMEM67 gene by Anastasiya Aleksandrovna Kozina, Guria Kurbanovna Kanaeva, Natalia Vladimirovna Baryshnikova, Anna Yurievna Ilinskaya, Anna Alexandrovna Kim, Anastasia Vladimirovna Erofeeva, Nadezhda Andreevna Pogodina, Jamilya Payzutdinova Gadzhiyeva, Ekaterina Ivanovna Surkova and Valery Vladimirovich Ilinsky in Journal of International Medical Research
Footnotes
Acknowledgements
The authors gratefully acknowledge and thank the proband and her family for their participation in the study.
Author contributions
AK, GK, NB, AI, AK, AE, NP, JG, ES, and VI met the International Committee of Medical Journal Editors criteria for authorship. AK, NB, AI, NP, ES, and VI contributed to the conception and design of this study. AK, GK, JG, NB, and ES collected, analyzed, and interpreted the clinical data. AI, AK, AE, and NP contributed to sequencing data collection and carried out the mutation analysis. AK, AE, JG, and ES drafted the manuscript. AK, GK, NB, AI, NP, and VI critically revised the manuscript. VI guided the completion of this manuscript and supervised and revised the manuscript for intellectual content. All authors read and approved the final manuscript.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request. The novel compound heterozygous variants were approved by ClinVar (VCV001801712.1, VCV000363927.11).
Declaration of conflicting interest
The authors declare no potential conflict of interest.
Ethics statement
The study was approved by the ethics committee of Genotek Ltd. (No. 09/2022). The patient's parents provided written informed consent for the study participation and publication of clinical information and sequencing data in this manuscript.
Funding
This research was funded by the Ministry of Science and Higher Education of the Russian Federation within the framework of state support for the creation and development of World-Class Research Centers “Digital Biodesign and Personalized Healthcare”, grant number 75-15-2022-305.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.