
Abstract
Fibrosing interstitial lung diseases (FILDs) other than idiopathic pulmonary fibrosis (IPF) can develop into progressive pulmonary fibrosis (PPF) despite initial management. A substantial proportion of patients with non-IPF interstitial lung diseases (ILDs) progress to PPF, including connective tissue disease-associated ILD (such as rheumatoid arthritis-associated ILD, systemic sclerosis-associated ILD, and idiopathic inflammatory myositis-associated ILD), fibrosing hypersensitivity pneumonitis, and fibrosing occupational ILD. The concept of PPF emerged only recently and several studies have confirmed the impact of PPF on mortality. In addition to poor prognosis among patients with PPF, there remains a lack of consensus in the diagnosis and treatment of PPF across different types of ILDs. There is a need to raise awareness of PPF in FILDs and to explore measures to improve PPF diagnosis and treatment, which in turn could potentially reduce the progression from FILD to PPF. This review discusses the disease burden of PPF and recent advances in the management of PPF among patients with ILDs, including antifibrotic medications that have emerged as promising treatment options. Additionally, this review highlights the perspectives of expert Chinese physicians with regard to their experience in managing PPF in clinical practice.
Keywords
Introduction
Fibrosing interstitial lung diseases (FILDs) are a heterogeneous group of diseases characterized by excessive deposition of extracellular matrix components within the pulmonary interstitium, leading to architectural distortion, irreversible pulmonary dysfunction, and early mortality. 1 These changes can be observed as reticular abnormalities, honeycombing, and traction bronchiectasis on high-resolution computed tomography (HRCT). 1 FILDs comprise idiopathic pulmonary fibrosis (IPF) and non-IPF FILDs. IPF is the prototypic progressive FILD1,2 and is defined as a chronic, fibrosing interstitial pneumonia with an unknown cause that presents with the radiological and histologic features of usual interstitial pneumonia (UIP), where UIP is characterized by the appearance of patchy dense fibrosis at low magnification that causes remodeling of the lung architecture, often resulting in honeycomb change, and alternates with areas of less-affected parenchyma.3,4 Non-IPF FILDs may also develop a progressive fibrosing phenotype despite appropriate treatment and are termed collectively as progressive pulmonary fibrosis (PPF), which is defined separately from IPF.1–3 The recommended management approaches for non-IPF FILDs may differ from those for PPF; immunosuppressive agents are the mainstay of treatment in non-IPF FILDs, whereas the progressive nature of PPF may limit the efficacy of standard-of-care immunosuppressive treatments.2,3,5
In addition to PPF, other similar terms are also commonly used, including progressive fibrosing ILD (PF-ILD) and chronic fibrosing ILD with a progressive phenotype. From a broad perspective, these three terms all describe similar groups of patients whose pulmonary fibrosis is progressively deteriorating despite proper management. Based on the recommendation in the 2022 American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Asociación Latinoamericana de Tórax (ATS/ERS/JRS/ALAT) clinical practice guidelines, PPF should be regarded as the preferred term to describe the disease. 3
Identification of PPF
Expert consensus indicates that HRCT scans are generally adequate for the diagnosis of non-IPF FILDs and for assessing disease progression. 6 However, in some cases, understanding the histological phenotype may be useful in determining the risk of progression when imaging is inconclusive. 6 In such cases, surgical lung biopsy can be considered where the benefits outweigh the potential risks; diagnostic transbronchial cryobiopsy or lung biopsy may be a safer alternative in certain patients. 6
In contrast, PPF is not considered a diagnosis, and the criteria for PPF are associated with prognosis. 3 The 2022 ATS/ERS/JRS/ALAT guidelines define PPF as a condition that meets at least two of the following three criteria occurring within the past year with no alternative explanation in patients with non-IPF ILD who have radiological evidence of FILD on HRCT: (1) worsening symptoms; (2) radiological progression; (3) physiological progression (Table 1). 3 The updated definition of PPF reflects the criteria used across different clinical trials that evaluated antifibrotic therapies; the criteria for PPF have been associated only with prognosis and may not identify patients who could benefit from antifibrotic therapy. 3 Rather than being a diagnosis, the definition of PPF is agnostic to the underlying condition. 3 The concept of PPF emerged only recently and the INBUILD trial, which included patients with a broad range of FILDs, provided evidence of PPF across different subtypes of ILD. 7 In the INBUILD trial, patients who received nintedanib, an intracellular inhibitor of receptor tyrosine kinases involved in the pathogenesis of ILDs, 8 had a slower progression of ILD than those who received placebo, reflected by a lower annual rate of decline in forced vital capacity (FVC) over the 52-week study period both in the overall trial population (−80.8 mL/year vs −187.8 mL/year; p < 0.001) and in patients with a UIP-like fibrotic pattern on HRCT (–82.9 mL/year vs −211.1 mL/year; p < 0.001). 7 Since then, several studies have evaluated the prevalence of PPF among non-IPF ILDs and confirmed the negative impact of PPF on lung function, which predicted mortality.9–11
Definition of PPF per ATS/ERS/JRS/ALAT clinical practice guidelines. 3

PPF is defined as at least two of the three criteria occurring within the past year with no alternative explanation. Although it is critical to exclude alternative explanations of worsening features for all patients with suspected progression, this is particularly important in patients with worsening respiratory symptoms and/or decline in DLCO given the lower specificity of these features for PPF compared with FVC and chest computed tomography.
⩾1 of the following evidence.
Either of the following evidence.
ALAT, Asociación Latinoamericana de Tórax; ATS, American Thoracic Society; DLCO; diffusing capacity of the lungs for carbon monoxide; ERS, European Respiratory Society; FVC, forced vital capacity; Hb, hemoglobin; JRS, Japanese Respiratory Society; PPF, progressive pulmonary fibrosis.
While the definition of PPF in the 2022 ATS/ERS/JRS/ALAT guidelines 3 and that in the INBUILD study trial 7 are largely aligned with only minor differences, such as progression of ILD over 24 months with a relative decline in FVC of > 10% predicted in the INBUILD trial versus 12 months with an absolute decline in FVC of ⩾5% predicted in the guidelines, physicians in real-world practice may have differing understanding of the concept of PPF. A study was conducted across three centers in the United States (US) to verify the prognostic value of the PPF criteria beyond categorical decline in FVC. 12 The study highlighted that an FVC decline of ⩾10% and six additional PPF criteria (combined with FVC, diffusing capacity of the lungs for carbon monoxide (DLCO), and HRCT) that are satisfied in the absence of categorical decline in FVC can identify patients with non-IPF ILD at increased risk for death or lung transplantation. 12 An expert group consensus statement involving physicians from different countries, including India and Europe, stated that although a 10% reduction in FVC was used as a stand-alone inclusion criterion in the INBUILD trial, smaller FVC reductions (5%–10%) associated with symptomatic or radiological deterioration were alternative inclusion criteria for the INBUILD trial.6,7 The value of prognosis assessment varies with different PPF criteria. 6 This consensus statement also demonstrated that several different terms have been used to describe the progressive fibrotic phenotype; however, none were applicable to the subgroup of non-IPF patients who were evaluated in recent antifibrotic trials as these terms omit the defining feature of progression of pulmonary fibrosis despite appropriate management. Specifically, “PPF” differs from “PPF despite appropriate management”. 6 In an online survey of 486 physicians, it was estimated that 18%–32% of patients with non-IPF ILDs develop PPF despite initial treatment, and the estimated time from detection of PPF to death in patients with non-IPF ILD was 30–45 months. 13 Of note, reduced lung function (decline in FVC or DLCO) has been shown to be a predictor of mortality.3,6 Thus, treatment that slows lung function decline should be considered for ILDs that exhibit a progressive fibrotic phenotype. 3
Patients with ILD and multiple underlying diseases can progress to PPF, including patients with (1) connective tissue disease-associated ILD (CTD-ILD), such as rheumatoid arthritis (RA)-associated ILD (RA-ILD), systemic sclerosis (SSc)-associated ILD (SSc-ILD), and idiopathic inflammatory myositis (IIM)-associated ILD (IIM-ILD); (2) fibrosing hypersensitivity pneumonitis (fHP), and; (3) fibrosing occupational ILDs.1,2,13 The prognosis of most patients with PPF is poor 14 ; in a cohort study of 150 patients with CTD-ILD, 76 patients (50.7%) developed PPF and this was associated with poor overall survival (adjusted hazard ratio 5.73, 95% CI 1.17–28.11). 15 In addition, there are several important clinical challenges in terms of the need for more effective medications, as well as diagnostic and/or prognostic biomarkers for PPF.16,17 The 2018 Chinese expert-based consensus statement for the diagnosis and treatment of CTD-ILD highlighted the need for increased attention to PPF in CTD-ILDs and for a consensus on the management of PPF in China. 18 Patient-reported outcomes indicate that patients with FILD who develop PPF experience significantly worsened quality of life, with increased symptom, activity, and impact scores as measured by the St George’s Respiratory Questionnaire (SGRQ), compared with those without PPF. 19 Furthermore, PPF was associated with clinically important deterioration in total scores of the SGRQ, chronic obstructive pulmonary disease assessment test, and 6-minute walking distance over two years, highlighting the increased disease burden in these patients. 19 This review will focus on the disease burden of PPF, the relevant treatment options for each ILD subtype, and recent advances in the management of PPF among Chinese patients with FILDs, highlighting the unmet needs in China and future perspectives in disease management.
PPF in CTD-ILD
Based on a 2017 online survey of physicians from France, Germany, Italy, Japan, Spain, the United Kingdom, and the United States, CTD-ILDs that were most likely to develop PPF included RA-ILD and SSc-ILD, accounting for 26% and 31% of patients with non-IPF ILDs, respectively. 13 In addition, patients with mixed connective tissue disease, IIM-ILDs, Sjögrens’ syndrome, vasculitis, and systemic lupus erythematosus are also likely to develop PPF. 3 A recent retrospective study of 307 Chinese patients with ILD, including those with CTD-ILD (120/307; 39%), RA-ILD (28/307; 9%), and SSc-ILD (26/307; 8%), found that combined pneumonia (odds ratio 4.57, 95% CI 2.54–18.43), low baseline DLCO% predicted (odds ratio 0.91, 95% CI 0.89–0.93), high modified Medical Research Council dyspnea score (odds ratio 6.34, 95% CI 4.23–10.14), and high HRCT score (odds ratio 1.38, 95% CI 1.25–1.54) were independent risk factors associated with progression to PPF (all p < 0.01). 20
Currently, low-dose glucocorticoids as monotherapy or in combination with immunosuppressive agents are used as first-line treatment in patients diagnosed with CTD-ILDs. 21 Commonly used immunosuppressants for the treatment of CTD-ILDs include cyclophosphamide, mycophenolate, azathioprine, and calcineurin inhibitors, with leflunomide also used in RA-ILD. 21 However, with the exception of cyclophosphamide and mycophenolate in SSc-ILD, their efficacy has not been validated in randomized controlled trials, and their use has been based on clinical experience and real-world data. 21 In patients who progress despite treatment with glucocorticoids and immunosuppressants, rescue therapy with biologic agents (such as rituximab, tocilizumab, or abatacept) can be considered, particularly in patients with ILDs with inflammatory involvement, or antifibrotic treatments. 21
Several recent clinical trials have shown that antifibrotic therapies can help slow disease progression in patients with ILD who have progressive fibrosing phenotypes.22–25 In 2014, the US Food and Drug Administration (FDA) approved the use of two oral antifibrotic medications, pirfenidone and nintedanib, in patients with IPF.8,26 Nintedanib has also been approved for the treatment of chronic FILD with a progressive phenotype and SSc-ILD, 8 supported by the findings of INBUILD, which demonstrated a significant slowing of FVC decline in patients treated with nintedanib versus placebo (between-group difference 107.0 mL/year, 95% CI 65.4–148.5, p < 0.001). 7 Both pirfenidone and nintedanib have been included in the ATS/ERS/JRS/ALAT guidelines for the management of CTD-ILDs, 3 and the relevant supporting data are described below.
(1) PPF in RA-ILD
RA is a chronic systemic autoimmune disease that affects approximately 5 million individuals in China according to data from 2021. 27 ILD is a major pulmonary complication of RA28,29 that has a significant impact on patients’ morbidity and mortality. 30 The majority of patients with RA-ILD have a restrictive pattern on pulmonary function tests and have poorer prognoses than other patients with RA due to the impairment of both FVC and DLCO. 31 The median survival of patients with RA after a diagnosis of ILD was 2.6 years, which was significantly lower than the expected median survival of 9.9 years in patients with RA of the same age and sex without ILD (p < 0.001). 32 In a retrospective study of 544 Chinese patients with RA, 83 (15.3%) patients were diagnosed with ILD based on HRCT; age at RA onset, elevated anti-cyclic citrullinated peptide, and steroid use were associated with RA-ILD. 33 These findings aligned with a recent meta-analysis of 56 studies of RA-ILD, which identified a pooled prevalence within the RA population of 18.7%, although significant between-studies heterogeneity should be noted. 34 Globally, it is estimated that 26% of patients with RA-ILD develop PPF. 13 While it remains challenging to identify the initiation of PPF in RA-ILD, several factors are associated with the development of PPF among patients with RA-ILD, including infection, smoking, immune response (e.g., Th-17-cell-mediated immunity and anti-citrullinated protein antibodies), genetics (e.g., mutations in telomere-related genes and the gene encoding mucin 5B), and premature senescence. 35
Patients with RA-ILD who progressed to PPF had lower peripheral platelet counts than those who did not progress to PPF, as demonstrated in a retrospective study. 36 Studies have also shown that patients with RA-ILD and the UIP pattern were more likely to develop PPF37,38 and had worse survival 39 than those with the non-specific interstitial pneumonia (NSIP) pattern. The UIP pattern in patients who are more likely to develop PPF is characterized by HRCT features such as heterogenous honeycombing of the pulmonary bases and periphery, peripheral basilar predominant reticular abnormalities, architectural distortions, anterior upper lobe honeycombing, and exuberant honeycombing. 40 Prognostic factors associated with the progression of RA-ILD (which includes the development of PPF) are summarized in Table 2. 21 Identifying prognostic factors may help physicians with earlier diagnosis of PPF in patients with RA-ILD and provide intensive targeted treatments and close surveillance for these patients.
Prognostic factors associated with the progression of CTD-ILDs, fHP, and fibrosing occupational ILDs.
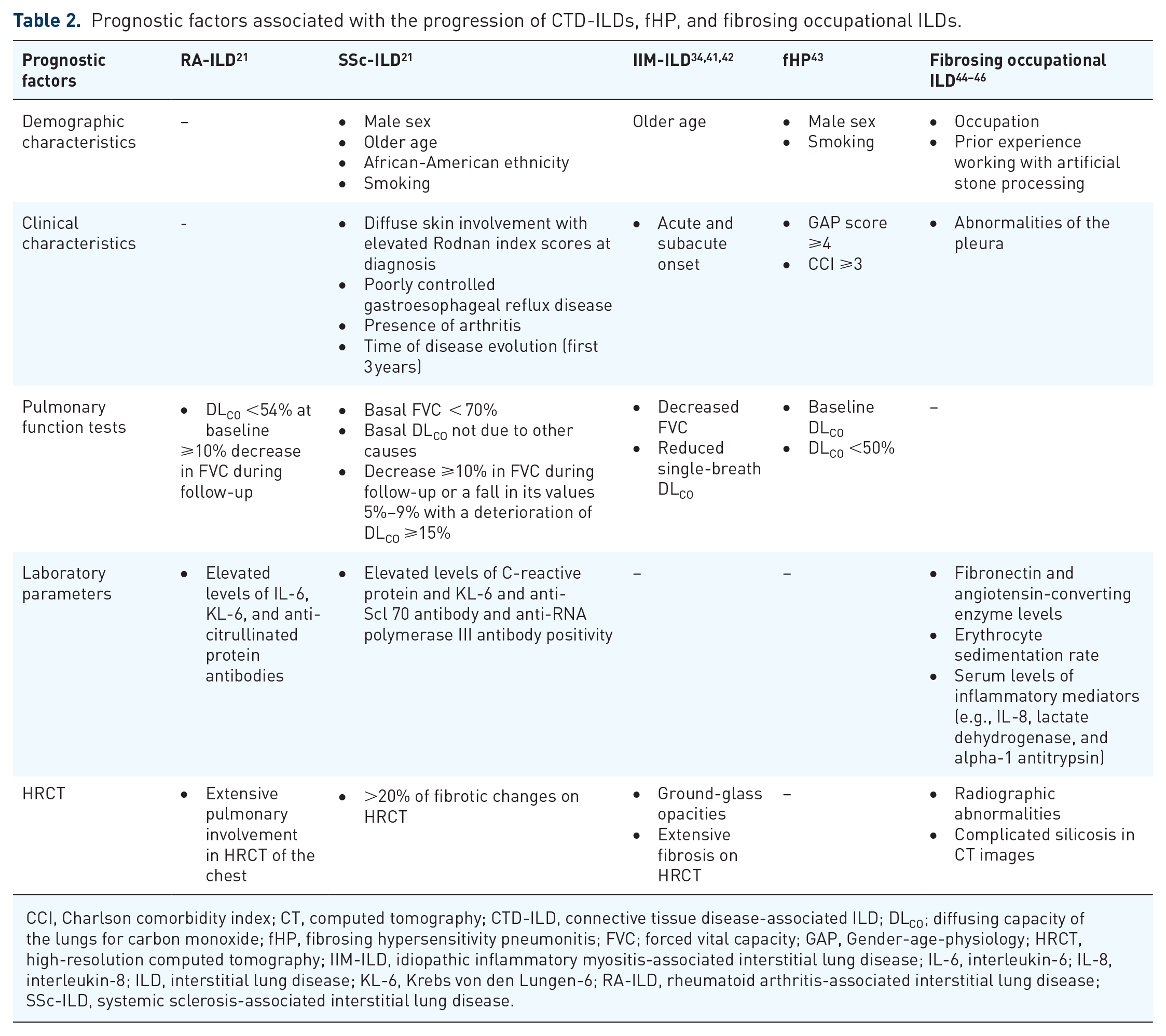
CCI, Charlson comorbidity index; CT, computed tomography; CTD-ILD, connective tissue disease-associated ILD; DLCO; diffusing capacity of the lungs for carbon monoxide; fHP, fibrosing hypersensitivity pneumonitis; FVC; forced vital capacity; GAP, Gender-age-physiology; HRCT, high-resolution computed tomography; IIM-ILD, idiopathic inflammatory myositis-associated interstitial lung disease; IL-6, interleukin-6; IL-8, interleukin-8; ILD, interstitial lung disease; KL-6, Krebs von den Lungen-6; RA-ILD, rheumatoid arthritis-associated interstitial lung disease; SSc-ILD, systemic sclerosis-associated interstitial lung disease.
In addition to glucocorticoids and immunosuppressant agents, methotrexate is another potential therapy for patients with RA-ILD. Historically, methotrexate had been discouraged in these patients due to the risk of acute pneumonitis, but randomized controlled trials have revealed that the risk is minimal (0.3% of patients treated with low-dose methotrexate).21,47 Furthermore, methotrexate use in patients with RA-ILD was associated with improved survival in a retrospective study in Mexico. 48 Antifibrotic agents can also be used in patients with RA-ILD. In a subgroup of 89 patients with RA-ILD who developed PPF in the landmark phase III INBUILD trial, there was a significant reduction in the rate of FVC decline over 52 weeks in the nintedanib group versus placebo (−82.6 mL/year vs −199.3 mL/year; p = 0.037), which was consistent with findings from the overall trial population that included patients with non-IPF ILDs who developed PPF. 22 In the phase II TRAIL1 trial that included 123 patients with RA-ILD, although pirfenidone slowed the decline in FVC, there was no statistically significant difference in the composite primary endpoint of decline in FVC% from baseline of ⩾10% or death in the pirfenidone versus placebo groups (odds ratio 0.67, 95% CI 0.22–2.03, p = 0.48). 49 However, these results should be interpreted with caution as the study was terminated early due to slow enrollment and the shutdown of routine clinical and research operational activities as a consequence of the coronavirus 2019 pandemic. 49
(2) PPF in SSc-ILD
SSc is a rare, heterogeneous connective tissue disorder characterized by microvascular damage, dysregulation of innate and adaptive immunity, and generalized fibrosis in the skin and multiple internal organs, including the lungs.50,51 Around one-quarter of patients develop significant pulmonary involvement within 3 years of SSc diagnosis, 52 with ILD being one of the most common forms of direct pulmonary involvement and a major cause of morbidity and mortality in patients with SSc.51,53 Globally, it is estimated that 31% of patients with SSc-ILD will progress to PPF. 13 Although little is known about how the mechanisms driving PPF differ across ILD subtypes, the potential mechanisms underlying the development of PPF in SSc-ILD is one of the most studied among the different ILD subtypes. 35 Persistent epithelial lung injury, endothelial cell dysfunction, and dysfunction in the innate and adaptive immune systems may contribute to the development of PPF. 35 The activation of macrophages may also be involved in the development of PPF in SSc-ILD 35 ; an exploratory analysis of 212 patients with SSc-ILD showed that elevated interleukin-6 levels may be predictive of mortality and significant lung function decline (FVC ⩾10% and DLCO ⩾15%). 54
NSIP pattern is the most common radiological morphological pattern in patients with SSc-ILD. However, radiographic patterns have not been shown to be associated with PPF in SSc-ILD. 35 This suggests that patients with non-IPF ILDs who develop PPF share some similarities, although differences also exist. SSc-ILD has a variable natural history with some patients having relatively stable disease for some time, while others develop PPF characterized by worsening fibrosis on HRCT, progressive decline in lung function, worsening dyspnea, and increased mortality.55–58 Prognostic factors associated with PPF in SSc-ILD are summarized in Table 2. 21 These prognostic factors may help physicians identify patients with SSc-ILD who are at risk of PPF development, which may allow appropriate, intensive targeted treatment to be implemented and facilitate their close surveillance in clinical practice.
Several studies have also explored the antifibrotic effects of pirfenidone and nintedanib in SSc-ILD.23,25,59 In a subgroup analysis of 170 patients with autoimmune disease-related ILDs who developed PPF in the INBUILD trial, 39 patients (22.9%) had SSc-ILD, in whom there was a slower rate of FVC decline over 52 weeks (primary endpoint) in the nintedanib group versus placebo (–75.9 mL/year vs −178.6 mL/year; p = 0.0117). 23 In the RELIEF trial that included 127 patients with CTD-ILDs (including SSc-ILD) who developed PPF and were not responsive to conventional treatment, there was a statistically significant difference in the primary endpoint of change from baseline in FVC% at Week 48 in the pirfenidone group versus placebo (difference 1.69, 95% CI −0.65 to 4.03, p = 0.043), although the results should be interpreted with caution due to premature study termination. 25 In the SENSCIS trial that included 576 patients with SSc-ILD, the primary endpoint of adjusted annual rate of change in FVC over a 52-week period was lower in the nintedanib group than in the placebo group (−52.4 mL/year vs −93.3 mL/year, difference 41.0 mL/year, 95% CI 2.9–79.0; p = 0.04). 59 Based on these studies, the 2024 ATS guidelines on the management of SSc-ILD suggest the use of nintedanib to treat patients with SSc-ILD, while further research into the safety and efficacy of pirfenidone was recommended. 60
In addition to antifibrotics, the ATS provided guidance on the use of immunosuppressive agents for the management of SSc-ILD, recommending the use of mycophenolate and suggesting the use of cyclophosphamide, rituximab, or tocilizumab. 60 Furthermore, the ATS guidelines suggest the use of nintedanib plus mycophenolate, based on findings from the SENSCIS trial, which found that when compared with placebo, patients treated with nintedanib plus mycophenolate had a reduced annual rate of decline in FVC and FVC% predicted, as well as a reduced risk of absolute decrease from baseline in FVC of > 5% and > 10% predicted. However, no significant differences were identified in the annual rate of FVC or FVC% predicted, nor were there differences in modified Rodnan skin score. 60 Further research into the use of pirfenidone plus mycophenolate is recommended by the ATS guidelines. 60 It should be noted that the ATS did not come to a consensus on a preferential or stepwise approach to these treatment interventions due to a lack of evidence, and the guidelines highlighted the evidence gaps that require further research before more prescriptive guidance could be provided. 60
(3) PPF in IIM-ILD
IIM is a heterogeneous group of diseases characterized by inflammation of the skeletal muscles and extra-muscular organs, including the lungs. 61 The most common clinical subtypes in adults are polymyositis and dermatomyositis. 62 ILD is one of the most important prognostic factors associated with poor survival in patients with IIM; 10-year survival is significantly worse in those with ILD versus those without ILD (66.1% vs 86.8%, p < 0.001) for Chinese adult patients with polymyositis or dermatomyositis. 63 Globally, it is estimated that 24% of patients with CTD-ILDs other than RA-ILD and SSc-ILD develop PPF. 13 However, the prevalence of PPF in patients with IIM-ILD remains unclear. 27 It is well established that anti-tRNA-synthetase antibodies, MDA5 and Ro52, are associated with the development of PPF in patients with IIM-ILD. 41 In addition, a recent study of 72 patients with anti-synthetase syndrome-associated (ASS) ILD in China found 18 (25%) patients classified as having PPF; multivariate logistic regression analysis identified positive non-Jo-1 antibodies, neutrophil-to-lymphocyte ratio, and serum KL-6 as independent risk factors for PPF development in patients with ASS-ILD. 64 However, the underlying pathogenetic mechanisms remain unknown.
NSIP, with or without overlapping organizing pneumonia (OP) or fibrosing OP pattern, is the most common radiological pattern in patients with subacute to chronic IIM-ILD. 65 The UIP pattern in IIM-ILD is typically an end-stage pathology of the NSIP and/or overlapping OP pattern and has been associated with an increased risk of PPF development.41,65 Diffuse cranio-caudal distribution and ground-glass opacities with random distribution may be associated with a rapid progressive course in myositis-associated ILD. 65 A retrospective study of 307 patients with IIM-ILD in China, of whom 94 (30.6%) had PPF, determined the median progression time from diagnosis to PPF was 7.5 months (range 0.9–12.0 months). 34 Multivariate Cox regression analysis revealed acute/subacute onset of pneumonia (hazard ratio 3.23, 95% CI 1.94–5.39), lower single-breath DLCO % predicted (hazard ratio 6.44, 95% CI 4.07–10.02), and the presence of diffuse alveolar damage on HRCT (hazard ratio 8.68, 95% CI 1.974–38.16) were associated with increased risk of progression to PPF (all p < 0.01). In contrast, patients receiving triple therapy had a reduced risk of developing PPF (hazard ratio 0.32, 95% CI 0.12–0.90; p = 0.031). 34 These and other proposed prognostic factors are summarized in Table 234,41,42; however, it should be noted that there are no established risk factors for PPF in patients with IIM-ILD.
PPF in fHP
Hypersensitivity pneumonitis (HP) is an immune-mediated ILD caused by inhalational exposure to low-molecular-weight compounds and may present in an acute, predominantly inflammatory form (non-fibrotic HP) or a chronic, fibrotic form (fHP). 66 Globally, it is estimated that 21% of patients with fHP develop PPF. 13 The pathogenesis of PPF in fHP remains unclear, although mutations in telomere-related genes and the mucin 5B gene have been implicated. 35 In fHP, short telomere length has been associated with extensive fibrosis, UIP pattern, and reduced survival, suggesting that fHP may have a common pathobiology with IPF. 67 Similar to RA-ILD, UIP is the predominant radiographic pattern associated with rapid disease progression in patients with fHP. 35 PPF in fHP has been associated with increased mortality. 43 Based on an analysis of registry data involving 292 patients with fHP, the group with the progressive phenotype had a longer duration of smoking, a higher gender, age, and physiology (GAP) score and Charlson comorbidity index, reduced lung function, a greater proportion of males (54.3% vs 41.5%), and more patients with comorbidities such as arterial hypertension (66.3% vs 53%), diabetes mellitus (23.9% vs 11%), and cancers (6.5% vs 1%), versus the group with nonprogressive phenotype. 43 Prognostic factors associated with PPF in fHP are summarized in Table 2. 43
Patients with fHP who progressed to PPF and/or were not responsive to conventional treatment may benefit from antifibrotic medications such as pirfenidone and nintedanib. There were 173 patients (26% of the total number of patients enrolled in the study) with fHP in the INBUILD trial. 24 Subgroup analysis showed that nintedanib reduced the rate of FVC decline when compared with placebo (difference in rate of FVC decline: 73.1, 95% CI −8.6 to 154.8, p = 0.41), although this difference was not statistically significant. 24 Furthermore, no new adverse effects were reported. 24 In a randomized controlled trial that included a small group of patients with chronic progressive fHP (N = 40), conventional treatment with add-on pirfenidone was associated with statistically significantly higher FVC and improved health-related quality of life (p < 0.001). 68 Pirfenidone also slowed FVC decline in patients with fHP in the RELIEF study. 25 Conversely, pirfenidone did not slow FVC decline compared with placebo in patients with fHP in another randomized clinical trial, although the study was underpowered to detect differences in the primary endpoint (mean absolute change in FVC%); no treatment-emergent serious adverse events were reported. 69
PPF in fibrosing occupational ILD
fHPs that result from workplace exposures are known as occupational fHP; occupational fHPs are acute or subacute in nature and usually result in ground-glass nodular opacities and mosaic attenuation on the HRCT. 70 Apart from occupational fHP, there are also fibrosing occupational ILDs, including asbestosis, silicosis, and chronic beryllium disease.13,70,71 Globally, it is estimated that 20–40% of patients with asbestosis would progress to PPF. 71 It was reported that 50% of patients with silicosis progressed to PPF in a retrospective Chinese cohort study. 33 The pathogenesis for progression to PPF in fibrosing occupational ILDs remains unclear, although it was reported that granuloma formation (in chronic beryllium disease) and persistent inflammatory response involving the generation of proinflammatory and profibrotic mediators (in asbestosis and silicosis) may be involved.35,71,72
Although nodular infiltrates are common on chest radiographs or HRCT scans, probable UIP pattern has been reported previously. 70 Radiologic manifestations for fibrosing occupational ILDs vary with exposure. 71 Restrictive pulmonary dysfunction has been observed in most fibrosing occupational ILDs.70,71 Reduced lung function, increased oxygen use, and increased number of acute hospitalizations may also be associated with the progression of PPF in occupational ILDs based on findings from one real-world single-center, retrospective study. 73 Prognostic factors for PPF patients with fibrosing occupational ILD are summarized in Table 2.44–46 Patients with fibrosing occupational ILDs who progressed to PPF and/or were not responsive to conventional treatment may benefit from antifibrotic medications such as pirfenidone and nintedanib.25,74–76 Nintedanib slowed the FVC decline in patients with exposure-related ILDs in an Asian subgroup analysis of the INBUILD trial, although the sample size for patients with exposure-related ILDs in this subgroup analysis was small (<7 patients). 75 Pirfenidone slowed the FVC decline in patients with fibrotic NSIP or asbestos-related PPF in the RELIEF study 25 and other studies,74,76 although the results from the RELIEF study should be interpreted with caution due to premature study termination. 25 There are several ongoing trials investigating the effects of nintedanib [ClinicalTrials.gov identifier: NCT04161014, NCT05067517] and pirfenidone [ClinicalTrials.gov identifier: NCT05118256, NCT04461587] in patients with fibrosing occupational ILDs who develop PPF.
Management of non-IPF ILDs
Guideline recommendations for the treatment of non-IPF ILDs manifesting as PPF
Following the FDA approval of antifibrotic medications in IPF, there has been increasing interest in better understanding how they may also slow the progression of non-IPF ILDs to PPF. The ATS/ERS/JRS/ALAT guidelines recommend nintedanib for the treatment of PPF in patients who have failed standard management for non-IPF ILDs manifesting PPF; further research on the efficacy and safety of pirfenidone in patients with non-IPF ILDs manifesting PPF is required. 3 The guidelines emphasize the need to identify those at risk of PPF who do not respond to initial treatment and may benefit from antifibrotic medications, and to prioritize research related to the timing and sequence of antifibrotic treatments with respect to corticosteroids and immunosuppressants for the various ILD subtypes that can manifest PPF. 3 In line with the ATS/ERS/JRS/ALAT guidelines, 3 the 2018 Chinese expert-based consensus statement 18 recommends that clinicians consider the likelihood of developing PPF when deciding the treatment paradigm for patients with CTD-ILD, with treatment goals of delaying disease progression and the development of PPF and improving patients’ quality of life. The 2023 American College of Rheumatology (ACR) guidelines conditionally recommend mycophenolate, rituximab, cyclophosphamide, and nintedanib as treatment options for individuals with systemic autoimmune rheumatic diseases-associated ILD progression despite initial ILD treatment, with additional conditional recommendations for and against the use of pirfenidone, tocilizumab, calcineurin inhibitor, JAK inhibitor, and intravenous immunoglobulin in different CTD-ILDs, summarized in Table 3. 77 However, currently, there is limited understanding of the optimal sequencing of these treatments, and the benefits of additional immunosuppressive agents versus earlier use of antifibrotics. Despite this limitation, these guidelines provide practical guidance for clinical use of antifibrotic drugs for the aforementioned patients.
2023 ACR recommendations for management of ILD progression despite first-line therapy.

Therapies are generally listed in order based on a hierarchy established by head-to-head votes by the ACR, but decisions should consider the specific clinical situation. Decisions on whether to switch or add therapy depend on current therapy and on which therapy is being initiated. Cyclophosphamide is typically not used in combination with other therapies. bThe choice of nintedanib versus immunosuppression depends on the rate of progression and the amount of fibrotic disease, or the presence of a UIP pattern on chest CT. cJAK inhibitor is conditionally recommended as an option, particularly in patients with anti-MDA-5 antibodies. dShort-term glucocorticoids may be of use in some patients with disease flares, or as a bridge when switching therapy.
ACR, American College of Rheumatology; ASCT, autologous stem cell transplant; IIM, idiopathic inflammatory myositis; ILD, interstitial lung disease; IVIG, intravenous immunoglobulin; JAK, Janus kinase; RA, rheumatoid arthritis; SSc, systemic sclerosis.
In patients with SSc-ILD, we recommend the treatment strategy outlined in the 2024 ATS guidelines, utilizing immunosuppressive therapy and antifibrotic agents. 60 Specifically, for immunosuppressive therapy, mycophenolate or cyclophosphamide should be considered the first choice, while for antifibrotics, nintedanib is recommended. Furthermore, we recommend a combination of immunosuppressive and antifibrotic therapy for the treatment of PPF in patients with SSc-ILD.
Management of acute exacerbations of FILD
It is important to distinguish between PPF and acute exacerbations of FILD due to the differences in disease course, clinical manifestations, and importantly, management. There is currently no official definition of an acute exacerbation of FILD in the setting of non-IPF ILDs; however, as these acute exacerbations resemble those in patients with IPF, clinically, the definition of acute exacerbations of IPF is applied. 78 Acute exacerbation of IPF has been defined as an acute, clinically significant respiratory deterioration of unidentifiable cause, and includes clinical worsening of less than a 30-day duration, new radiologic abnormalities on HRCT, and the exclusion of alternative etiologies, such as infection, or pulmonary embolism. 79
Currently, there are limited evidence-based data on effective treatment for acute exacerbations of FILD. Noninvasive ventilation can provide support by reducing breathlessness in patients with acute exacerbations of FILD; however, detailed discussions should be had with the patient prior to obtaining their consent. 6 Acute exacerbations of ILD are also often treated with high-dose systemic corticosteroid therapy plus antibiotics. 78 Observational studies have shown that immunosuppressive treatments, particularly combination therapy with corticosteroids, as well as rituximab, may be beneficial in acute exacerbations. 78
Due to the lack of effective treatment for acute exacerbations, prevention is an important therapeutic goal. Antifibrotic therapies may provide a protective effect against acute exacerbations, 6 with the INBUILD trial finding a reduced risk of acute exacerbation or death in patients treated with nintedanib compared with placebo (13.9% versus 19.6%, respectively; hazard ratio 0.67, 95% CI 0.46–0.98, nominal p = 0.04). 7 However, currently there are insufficient data to determine whether these therapies should be continued or paused during an acute exacerbation, 6 and further prospective studies are required to identify the most appropriate management options for acute exacerbation.
The role of transplant in PPF
We recommend consultation for transplant in patients with PPF whose disease progresses despite comprehensive treatment with active drug and nondrug therapy, with significant impairment in lung function to the level where transplant would be considered in patients with IPF, if the patient is otherwise fit without contraindications. In particular, patients with a probable or definite UIP pattern should be referred for transplant at the time of diagnosis. 80 Nondrug therapies, such as lung rehabilitation and oxygen therapy, are also important considerations in PPF, and the strategies in IPF can be referred to for patients with PPF, but each patient’s plans should be individualized and adjusted according to their disease condition.
Lung transplants may extend and improve the quality of life in patients with advanced lung diseases, and ILD has been the most common indication for lung transplantation since 2007, comprising 40.5% of lung transplants worldwide. 81 The International Society for Heart and Lung Transplantation recommends early referral for transplantation, particularly in patients with chronic lung disease, including ILD, who have a high risk of death in 2 years and a high likelihood of 5-year post-transplant survival. 80 Early referral also allows patients to better prepare for transplantation and address any issues that may complicate or delay transplantation, such as obesity, malnutrition, other medical comorbidities, or inadequate social support.80,82 While a number of risk factors are absolute contraindications to lung transplant, such as acute liver or kidney failure, it should be noted that old age is not an absolute contraindication, but older individuals tend to have worse long-term survival post-transplant, and patients should be considered on a case-by-case basis. 80
Where transplant is unavailable, or is contraindicated in the patient, the patient’s specific needs and goals of care should be considered when deciding on further management, including consideration of palliative care. 6
Chinese clinical perspective
In diagnosing PPF, we consider PPF to be a disease behavior and a clinical process of FILD, and utilize the diagnostic approach in the 2022 ATS/ERS/JRS/ALAT guidelines. 3 Future improvements in artificial intelligence may provide better precision and refinement in thoracic imaging to aid in the diagnosis of PPF. 83 Bronchoscopy is not routinely performed, except where serological screening reveals an infection; similarly, cryobiopsies are not regularly performed unless the patient has a combined tumor. 83
While we agree with expert consensus recommendations for early referral for a lung transplant, in our experience, transplant waiting times can be 6–8 weeks for patients with PPF. Thus, other nondrug treatments and multidisciplinary management play an important role in the management of PPF, including rehabilitation treatments such as oxygen therapy, as recommended in the Clinical Practice Guideline on Home Oxygen Therapy for Adults with Chronic Lung Disease. 84 Furthermore, traditional Chinese medicines have demonstrated efficacy in the improvement of symptoms associated with ILD; a recent meta-analysis of seven studies in patients with CTD-ILD found that combining traditional Chinese herbal medicine with cyclophosphamide significantly improved FVC, FEV1, total lung capacity, and DLCO, as well as reducing HRCT signals, when compared with cyclophosphamide alone, without an increase in adverse events. 85 However, it should be noted that different herbal medications were used in these studies, and the specific active components are not yet understood, but this remains a promising area of investigation.
Unmet needs and future perspectives
Given that there remains a lack of consensus in the diagnosis and treatment of PPF for different underlying diseases, there is a need to explore the pathogenesis of PPF and measures that could potentially prevent the progression of FILDs to PPF.
First, the diagnosis of PPF would need to be improved. 3 At present, any changes or progression of ILDs are evaluated based on worsening respiratory symptoms and changes in chest CT and lung function. 3 Although there are plasma biomarkers for PPF (e.g., elevated levels of Krebs von den Lungen-6, surfactant protein A and/or D, 16-kDa Clara cell secretory protein, and matrix metalloproteinase 1 and 7), 86 these have yet to be validated in prospective studies. 6 Furthermore, studies of self-reported measures and respiratory questionnaires (e.g., SGRQ and King’s Brief Interstitial Lung Disease Questionnaire) have been retrospective and limited by small sample sizes, and require further validation before they can be used as a prognostic indicator. 6 Proteomic analyses and transcriptomic studies may uncover serum biomarkers that can potentially identify patients at risk of PPF and predict disease progression. 3 Second, improved understanding of the best strategies for antifibrotic treatment, including timing, drug selection, dosage, and duration, is needed. 18 Antifibrotic medications have shown promising treatment effects for PPF in FILDs other than IPF 18 . The INBUILD trial demonstrated that nintedanib can slow the progression of ILD across a broad range of non-IPF ILDs with a progressive fibrosing phenotype3,7,41; the efficacy of pirfenidone in patients with non-IPF PPF remains unclear based on published studies (RELIEF, TRAIL1, and UILD).3,49 In addition to nintedanib and pirfenidone, other antifibrotic treatments are under investigation. Studies evaluating the safety and efficacy of BI 1015550 (a phosphodiesterase 4B inhibitor) [ClinicalTrials.gov identifier: NCT05321082], 87 BMS-986278 (a lysophosphatidic acid receptor 1 antagonist) [ClinicalTrials.gov identifier: NCT06025578], 88 treprostinil (a TREK-1/2 potassium channel antagonist) [ClinicalTrials.gov identifier: NCT05943535], 89 and pirfenidone (a structural analog of pirfenidone)[ClinicalTrials.gov identifier: NCT05139719] 90 in patients with progressive FILDs other than IPF are ongoing. Last but not least, more studies are required to better understand the pathogenesis of PPF in FILDs. 41 High-throughput genomics, proteomics, and metabolomics studies are required to identify the pathways underlying the progression of non-IPF FILDs to PPF. 35
For Chinese patients, there remains a lack of published studies on PPF development. As described above, retrospective studies in small populations have been published regarding the disease course of ILD to PPF; however, prospective studies would strengthen these findings.
Conclusion
The manifestation of PPF in non-IPF ILDs represents a significant disease burden and can pose a challenge to the clinical management of ILDs. It is crucial to raise awareness of PPF in ILDs and to standardize disease management strategies.