
Abstract
Background:
Idiopathic pulmonary fibrosis (IPF) is the most common idiopathic interstitial pneumonia and has a median survival after diagnosis of 2–5 years. Pirfenidone is the first approved antifibrotic drug for the treatment of IPF. Here we report the functional progress, side effects and survival data of a population of patients with IPF, diagnosed at our centre and treated with pirfenidone.
Methods:
We enrolled 91 patients with IPF (71 males) treated with pirfenidone. Clinical, survival and functional details were collected retrospectively at start of therapy and after 12, 24, 36 and 48 months of treatment. Lung function tests at least 12 months before starting therapy were available for 40 patients and were entered in the database, as well as side effects.
Results:
During the observation period (922 ± 529 days), 27 patients died, 5 patients underwent lung transplant and 10 patients interrupted therapy due to adverse events or IPF progression. The median survival was 1606 days. There was a significant reduction in disease progression rate, as measured by trend of forced vital capacity, after 1 year of therapy with respect to before treatment (p = 0.0085). Forced vital capacity reduction rate was progressively higher in the subsequent years of treatment. Treatment-related side effects were reported in 25 patients and were predominantly mild. Overall, four patients discontinued therapy due to severe photosensitivity.
Conclusions:
Our findings confirm the efficacy of pirfenidone in reducing functional progression of IPF and its excellent safety profile in a real-life setting. This study, designed on a long-term follow up, contributes to the growing evidence on safety, tolerability and efficacy of pirfenidone in IPF.
The reviews of this paper are available via the supplemental material section.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a debilitating interstitial lung disease (ILD) of unknown aetiology, limited to the lungs and characterized by usual interstitial pneumonia (UIP) radiological and histological pattern. 1 Morbidity and mortality are high: estimated 5-year survival is 20–40%.1–3 Its prevalence ranges from 1.25 to 23.4 cases per 100,000 of population 4 ; estimated incidence ranges from 2.8 to 19 cases per 100,000 persons/year. 5 The negative prognosis of IPF is related to the chronically progressive nature of the disease, leading to an irreversible impairment of lung volume and diffusion capacity that results in respiratory failure development and death. Moreover, IPF is often complicated by many respiratory comorbidities, such as pulmonary hypertension, obstructive sleep apnoea and lung cancer, that significantly worsen quality of life and life expectancy of these patients.6–8
The two drugs, nintedanib and pirfenidone, were recently approved for treatment of IPF. Both drugs have shown significant efficacy in reducing functional decline and have an acceptable safety profile, even though they are not capable of halting progression of disease. 9 Phase III clinical trials ASCEND, CAPACITY 004 and CAPACITY 006 were the first to report a significant beneficial effect of pirfenidone in reducing the decline of forced vital capacity (FVC) in patients with IPF.10,11 Subsequent open-label extension trials and real-life multicentre studies have confirmed the efficacy of pirfenidone in reducing disease progression,12–17 leading to a significant improvement in survival.18,19 Safety concerns are few, including specifically gastrointestinal symptoms and photosensitivity, and can be usually resolved or mitigated through a careful management of drug assumption. 9 The Italian Medicine Agency approved pirfenidone for clinical use in Italy in June 2013. Regional Referral Centres for ILDs can prescribe the drug for patients with early-to-moderate IPF, according to national drug inclusion/exclusion criteria: age 40–80 years, FVC ⩾ 50% of theoretical value and diffusing capacity of the lung for carbon monoxide (DLCO) ⩾ 35% of predicted value.
Here we evaluated the effectiveness and safety of pirfenidone in a population of 91 patients with IPF treated by our centre between 2011 and 2019. Our aim was to contribute to the definition of the long-term effectiveness and tolerability of pirfenidone in clinical practice.
Methods
Study population
A total of 91 patients with IPF (71 males, mean age 68.46 ± 7.70 years) treated with pirfenidone were enrolled retrospectively in the study, starting in 2011. From 2011 to June 2013, pirfenidone was only available for clinical trials, and subsequently also for compassionate use. All patients were diagnosed according to the American Thoracic Society (ATS)/European Respiratory Society (ERS) guidelines 1 at the Siena Referral Centre for ILDs.
Clinical and functional parameters were collected and entered in an electronical database: demographic data, family history for pulmonary fibrosis, smoking status, occupational/environmental exposure, functional parameters and comorbidities.
The pulmonary function tests (PFTs) of 40 patients (44%) were also available 1 year before starting pirfenidone therapy and were also recorded.
Radiological and immunological data were also available.
All patients underwent high-resolution computed tomography (HRCT) of the chest for diagnostic purposes. CT scans were interpreted by a radiologist experienced in ILDs and diagnosis of IPF was conducted in a multidisciplinary setting.
During follow up, clinical assessment and PFTs were performed every 3 months, according to the protocol of our centre. Clinical and functional data were entered in the database, together with treatment-related side effects. Survival data were also recorded. Functional disease progression was defined as a 1-year decrease of FVC > 10% or a 1-year decrease of DLCO > 15%, as previously suggested. 19
PFTs
The following lung function parameters were recorded according to ATS/ERS standards,20,21 using a Jaeger body plethysmograph with corrections for temperature and barometric pressure: forced expiratory volume in 1 s (FEV1), FVC, FEV1/FVC, total lung capacity (TLC), residual volume (RV), transfer factor of the lung for carbon monoxide (TLCO) and TLCO/alveolar volume (VA).
Statistical analysis
All data were expressed as the mean ± standard deviation, unless otherwise indicated. Statistical analysis was performed with GraphPad Prism version 5.0 software for Windows (GraphPad Software, La Jolla, CA, USA) using non-parametric tests. Spearman correlation index (r) was used to examine correlations between quantitative variables. Unadjusted survival and disease progression outcome estimates were obtained using Kaplan–Meier curves. Time-to-event endpoints were compared using a two-sided log-rank test. A p value ⩽0.05 was considered significant.
Results
All 91 patients enrolled were included in the analysis of this study. As expected, most of our patients were male (78%), over 65 years old (72.5%), current/former smokers (63%) and reported at least one medical comorbidity. Exertional dyspnoea and chronic dry cough were the main symptoms reported at onset. Baseline chest examination revealed bibasal crackles in all patients. The majority of patients showed a definite UIP pattern at HRCT of the chest, while emphysema was radiologically evident in 15 patients (Table 1).
Demographic features, smoking status and radiological parameters in IPF population at baseline. All data are expressed as mean ± standard deviation.
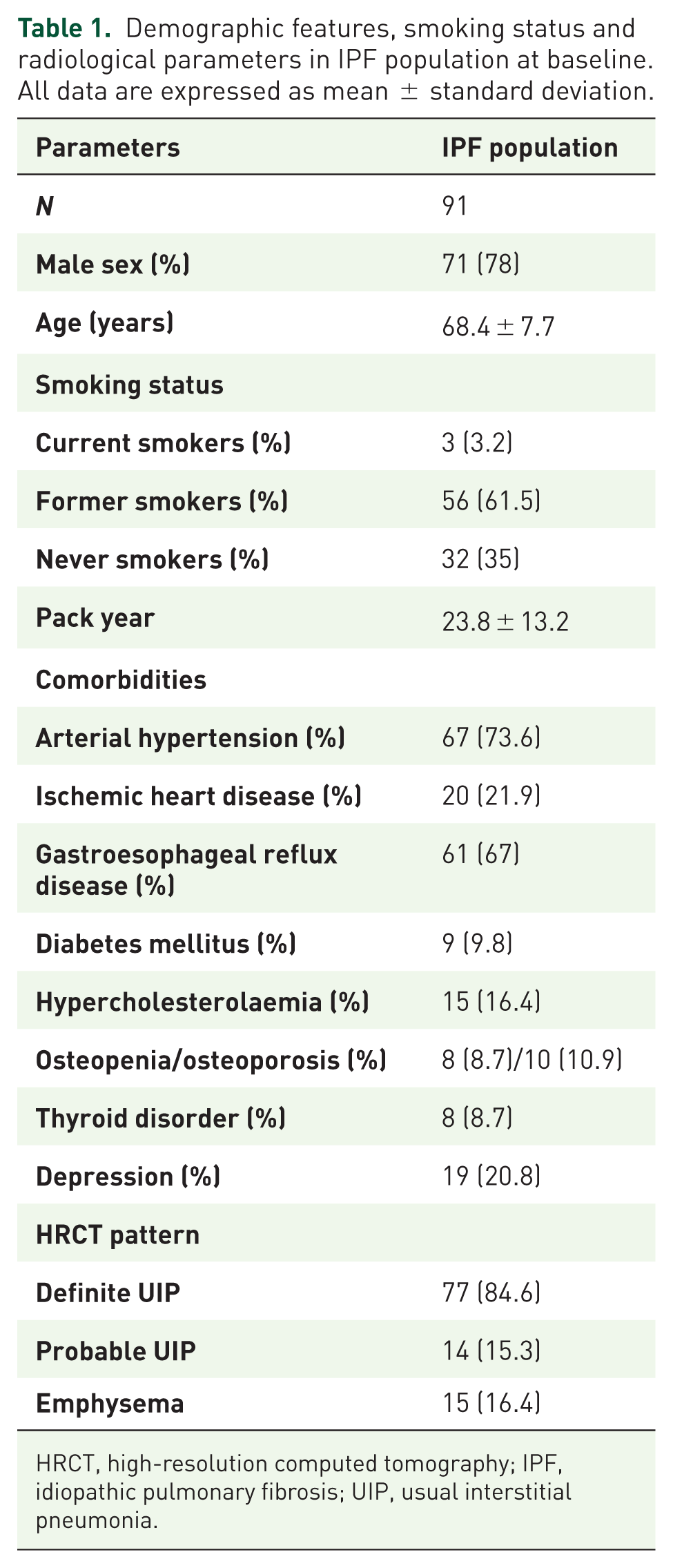
HRCT, high-resolution computed tomography; IPF, idiopathic pulmonary fibrosis; UIP, usual interstitial pneumonia.
Concerning functional parameters, at baseline a mild restrictive impairment and a moderate reduction of DLCO were observed in our population. Beyond baseline assessment, PFTs were available in 40, 75, 51, 27 and 18 patients, at 12 months before recruitment and after 12, 24, 36 and 48 months of therapy, respectively (Table 2).
Functional parameters of patients with IPF in pretreatment period, at baseline (initiation of pirfenidone therapy) and during follow up.

difference against baseline values.
DLCO, diffusing capacity of the lung for carbon monoxide; FVC, forced vital capacity; IPF, idiopathic pulmonary fibrosis.
Outcome analysis
Clinical follow up was available for all patients. On 1 July 2019 (after 922 ± 529 days of follow up), 28 patients had died (30.7%), 5 patients had undergone lung transplant (5.4%) and 48 patients (52.7%) were still alive and treated with pirfenidone. Overall, 10 patients (10.9%) changed from pirfenidone to nintedanib due to disease progression (6 patients, 821 ± 301 days of pirfenidone therapy) or severe side effects (4 patients, 492 ± 427 days of pirfenidone treatment). They were considered lost to follow up.
The overall percentage of patients showing a significant decrease in FVC (>10% with respect to baseline) at 12, 24, 36 and 48 months was 18% (14 of 75 patients), 41% (21 of 51), 55% (15 of 27) and 50% (9 of 18), respectively. The median time to decline in FVC > 10% was 449 days.
In the subgroup of patients with known pretreatment PFTs (40 patients, 44%), a significant decrease in FVC reduction rate (7.1% ± 6.7 versus 3.1% ± 7, U = 936.5 p = 0.0089; in absolute values, 226.3 ml ± 229.7 versus 86.8 ± 238.6, U = 876, p = 0.0085) was observed after 1 year of treatment (Figure 1). No significant differences in DLCO were observed (U = 989, p = 0.7185).

Comparison of FVC reduction in 40 patients with IPF 1 year before and 1 year after therapy with pirfenidone. (a) absolute values expressed as ml (**p = 0.0085); (b) percentage values (**p = 0.0089).
A significantly higher percentage of patients showed a FVC decrease > 10% in the pretreatment period compared with the first year of treatment (log-rank test 8.0059, p = 0.0045, hazard ratio of 3.569) (Figure 2).
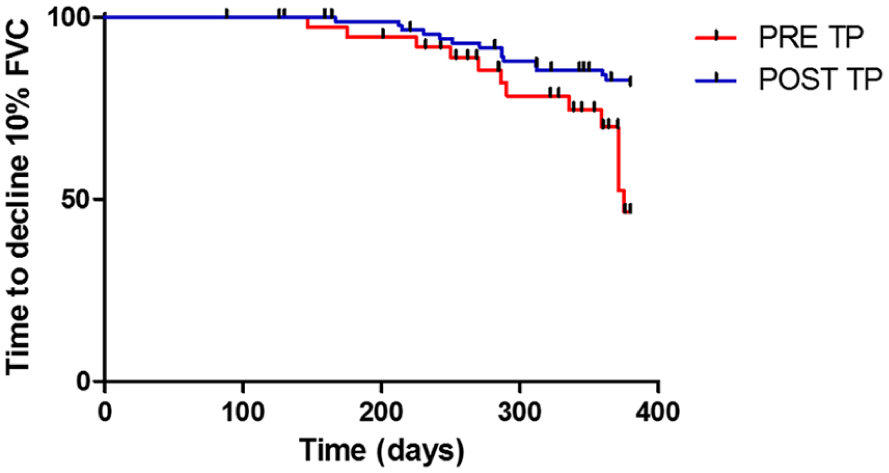
Kaplan–Meier curves comparing time to decline of 10% FVC in patients with IPF 1 year before and after pirfenidone therapy.
After an observation period (total 83 months), the median survival of our population was 1606 days. Acute exacerbation of IPF (AE-IPF) was diagnosed in seven patients (7.6%) and was fatal in all cases. Overall causes of death were IPF progression (17 patients, 60.7%), AE-IPF (7 patients, 25%), heart failure (2 patients, 7.1%), lung adenocarcinoma (1 patient, 3.5%) and septic shock (1 patient, 3.5%).
Safety
Pirfenidone-related side effects were reported in 25 of 91 patients (27%) (Table 3) and half of these appeared in the first 3 months of therapy. The most common side effect was skin rash (six patients, 6.5%). Only four patients (4%) permanently discontinued therapy due to drug intolerance: in all these cases, interruption was due to severe photosensitivity. Prokinetic drugs and proton-pump inhibitors were used to manage gastrointestinal side effects and broad-spectrum sunscreen was used to protect against skin reactions. Skin rash was severe in two cases due to persistent occupational sun exposure.
Adverse events reported in our cohort.

Excluding those who interrupted treatment, only 3 of 87 patients (3.4%) underwent temporary dose reductions or interruption due to cutaneous or gastrointestinal side effects (one and two patients, respectively).
Discussion
In this retrospective study, we describe our experience with pirfenidone in a cohort of patients with IPF treated at our referral centre for ILDs, focusing on functional disease progression, survival and drug safety. As expected, males over 65 years old were prevalent in our population with respect to females and younger patients; all had evidence of bibasal inspiratory crackles and a restrictive impairment at PFTs, associated with a reduction of DLCO.
Our results confirm the efficacy of pirfenidone in significantly slowing FVC decline, as reported by other authors in clinical trials9,10 and real-life observational studies.22,23 Mean decline in FVC was similar to those previously reported, further supporting the efficacy of pirfenidone in patients with IPF. In particular, the FVC reduction rate was more than halved after 1 year of treatment (from 210.5 ± 251.4 to 91.7 ± 237.4 ml), confirming good speed of action.
To our knowledge, this is the fourth study to investigate the efficacy of pirfenidone over an observation period greater than 2 years in a real-life setting. A progressive acceleration of FVC reduction rate after 24, 36 and 48 months of therapy was observed, although FVC remained lower in the pretreatment period. Bando and colleagues 24 and Tzouvelekis and colleagues 23 reported similar findings. On the other hand, an observational study based on the Czech IPF registry showed substantial stability of FVC progression rate even after 2 years of therapy. 14
The latter discrepancy may be due to the progressive decrease in our sample size, which also included patients with severe lung volume impairment at baseline. At the same time, we cannot exclude that pirfenidone prevented an even more pronounced decline in FVC, as reported by the metanalysis of Nathan and colleagues 19 Moreover, the RECAP study, an open-label extension of the CAPACITY and ASCEND clinical trials, showed an annualized rate of FVC decline of 144 ± 6 ml over a follow up of 180 weeks, which was similar to our findings. However, since RECAP was not specifically designed for the evaluation of long-term efficacy, a specific longitudinal study on a larger population is needed to clarify whether pirfenidone maintains its efficacy over a follow up of more than 1 year.
Concerning mortality, our data showed a median survival of 1606 days. Despite the absence of a control group, if compared to other historical cohorts available in literature,14,18,25 our data seem to confirm the efficacy of pirfenidone in prolonging life expectancy in patients with IPF. However, the survival time of our population was worse than that reported in the Czech registry and RECAP study.12,14 This discrepancy may be determined by inclusion in our study of a subgroup of patients with advanced IPF, who were taking pirfenidone on compassionate grounds. In fact, other real-life studies18,26 recruiting patients with severe disease, reported mortality data in line with ours. Moreover, our population showed a higher prevalence of medical comorbidities, such as arterial hypertension or gastroesophageal reflux disease, in respect to randomized controlled trials, RECAP and Czech registry studies,10–12,14 but comparable with other real-life studies. 18 These data may be influenced by previous steroid therapy, particularly in those whose IPF diagnosis had been made before 2012 or those using pirfenidone for compassionate use.
Interestingly, despite poorer survival data, our population showed fewer episodes of AE-IPF compared with other studies.13,26 These findings may be related to the difficulty of performing an accurate diagnosis of AE-IPF in a clinical setting; moreover, the definition of AE-IPF has changed in the last 10 years, and now also includes acute worsening of disease of known origin (e.g. infections). 27 This may be a confounding factor in reporting AE-IPF, especially in retrospective observational studies.
Our results confirm the good tolerability profile of pirfenidone. Only a small percentage of patients were forced to stop therapy because of incoercible photosensitivity. Side effects were predominantly mild to moderate and temporary dose reduction was only necessary in a minority of patients. We observed fewer side effects than in other real-life studies.19–22 We strongly encouraged use of sunscreens in all patients treated with pirfenidone and most patients were also on antacid therapy. Moreover, as suggested by Costabel and colleagues, 28 patients were accurately informed concerning the potential adverse effects of pirfenidone and were strongly encouraged to apply the preventive measures suggested; these factors may have contributed to better tolerance of the drug. No major liver toxicity was recorded in our cohort, further confirming the good tolerability profile of pirfenidone.
Considering the aim of this research, our study was subject to many limitations, specifically, its retrospective and monocentric nature is typically prone to recall and misclassification bias. Moreover, the absence of a control group did not allow us a more precise evaluation of pirfenidone efficacy in reducing the progression rate of disease.
In conclusion, the present study described our 8-year experience with pirfenidone in the treatment of patients with IPF. The drug was confirmed to be effective in significantly slowing functional progression of disease, leading to an increase in life expectancy. No new safety alert emerged, supporting the good tolerability profile of the drug. More data are needed to elucidate the effectiveness of pirfenidone in reducing the rate of AE-IPF and to clarify its long-term efficacy on disease progression. Studies investigating effectiveness of pirfenidone in AE-IPF and studies investigating its synergistic effect with novel compounds that entered the pipeline of clinical trials (i.e. pamrevlumab and pentraxin) are greatly anticipated29–31and will hopefully transform the therapeutic management of patients.
Supplemental Material
Author_response – Supplemental material for Pirfenidone in idiopathic pulmonary fibrosis: real-life experience in the referral centre of Siena
Supplemental material, Author_response for Pirfenidone in idiopathic pulmonary fibrosis: real-life experience in the referral centre of Siena by Lucia Vietri, Paolo Cameli, Marco Perruzza, Behar Cekorja, Laura Bergantini, Miriana d’Alessandro, Rosa Metella Refini, Maria Pieroni, Antonella Fossi, David Bennett, Marco Spalletti, Maria Antonietta Mazzei, Piersante Sestini, Paola Rottoli and Elena Bargagli in Therapeutic Advances in Respiratory Disease
Supplemental Material
Reviewer_1_v.1 – Supplemental material for Pirfenidone in idiopathic pulmonary fibrosis: real-life experience in the referral centre of Siena
Supplemental material, Reviewer_1_v.1 for Pirfenidone in idiopathic pulmonary fibrosis: real-life experience in the referral centre of Siena by Lucia Vietri, Paolo Cameli, Marco Perruzza, Behar Cekorja, Laura Bergantini, Miriana d’Alessandro, Rosa Metella Refini, Maria Pieroni, Antonella Fossi, David Bennett, Marco Spalletti, Maria Antonietta Mazzei, Piersante Sestini, Paola Rottoli and Elena Bargagli in Therapeutic Advances in Respiratory Disease
Supplemental Material
Reviewer_2_v.1 – Supplemental material for Pirfenidone in idiopathic pulmonary fibrosis: real-life experience in the referral centre of Siena
Supplemental material, Reviewer_2_v.1 for Pirfenidone in idiopathic pulmonary fibrosis: real-life experience in the referral centre of Siena by Lucia Vietri, Paolo Cameli, Marco Perruzza, Behar Cekorja, Laura Bergantini, Miriana d’Alessandro, Rosa Metella Refini, Maria Pieroni, Antonella Fossi, David Bennett, Marco Spalletti, Maria Antonietta Mazzei, Piersante Sestini, Paola Rottoli and Elena Bargagli in Therapeutic Advances in Respiratory Disease
Footnotes
Acknowledgements
The present research was performed at Siena University without funding sponsors. Lucia Vietri and Paolo Cameli contributed equally.
Author contributions
The author contributions were as follows: LV, PC, MP, EB and PS: conception and study design, revision of the study, interpretation of results. BC, LB, MDA, RMR, MP, AF, DB, MS, MAM and PR: data acquisition and analysis, revision of the study. PC, EL and PS: statistical analysis, revision of the study. All authors approved the final version of the study and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
The authors declare no conflicts of interest in preparing this article.
Supplementary material
The reviews of this paper are available via the supplemental material section.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.