
Abstract
Aclidinium is a potent and selective muscarinic antagonist, which interacts rapidly with muscarinic receptors and shows subnanomolar affinity for the five human muscarinic receptors (M1–M5); its association rate for the M3 receptor is similar to that of ipratropium and 2.6 times faster than that of tiotropium. Aclidinium dissociates slightly faster from M2 and M3 receptors than tiotropium but much more slowly than ipratropium. A potent bronchodilatory activity has been observed after inhaled administration of aclidinium. Aclidinium undergoes rapid hydrolysis in the plasma into two major compounds, the alcohol (LAS34823) and the carboxylic acid (LAS34850) metabolites, resulting in low and transient systemic exposure to the active drug. The two major metabolites show no significant affinity for human muscarinic receptors. A potent bronchodilatory activity has been observed after inhaled administration of aclidinium. Clinical trials have provided evidence of sustained bronchodilation similar to that observed with tiotropium. Trial results have confirmed the positive safety profile of aclidinium, particularly in terms of a very low propensity to cause anticholinergic adverse events. Aclidinium is now moving to phase III clinical development for chronic obstructive pulmonary disease (COPD).
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by irreversible airflow limitation associated with inflammation and structural changes of the lung and important comorbid conditions. These abnormalities may contribute to the natural history and severity of the disease in a given patient. It is estimated that COPD caused approximately 2.7 million deaths worldwide in 2002 [Lopez et al. 2006]. At present, COPD represented the sixth most common cause of mortality but the disease is expected to be the fourth most common cause of mortality worldwide by 2030 and the third most common cause of chronic disability by 2020 [Lopez et al. 2006]. This common chronic disease represents a major burden for patients, affecting their health-related quality of life and constitutes important costs for the society due to exacerbations, hospitalizations and work absenteeism. Cigarette smoking is the most commonly encountered risk factor for COPD in Europe and the USA, although in many other countries, domestic and outdoor air pollution, resulting from the burning of wood and other biomass fuels, have also been identified as potential COPD risk factors especially in nonsmoking women [Lopez et al. 2006]. Chronic airflow limitation is caused by a combination of inflammation of small airway diseases (obstructive bronchiolitis) and parenchymal destruction (emphysema). Chronic inflammation potentially causes structural changes and narrowing of small airways but the chronology of the events is not completely understood. Destruction of the lung parenchyma leads to the loss of alveolar attachments to the small airways and decreases lung elastic recoil; in turn, these changes diminish the ability of the airways to remain open during expiration in a so-called ‘dynamic hyperinflation’ mechanism [GOLD, 2009]. Exacerbations are mainly related to infections which contribute to accelerate decline of the lung function. COPD is generally a progressive disease linked to noxious agent exposure. Stopping exposure to these agents, even when significant airflow limitation is present, may result in some improvement in lung function. However, once developed, COPD cannot be cured and thus must be treated continuously. At present, apart from smoking cessation, there is no treatment able to interfere with the natural history of the disease. Modern pharmacological management has been shown to reduce symptoms, improve quality of life, reduce exacerbations and possibly reduce mortality [GOLD, 2009], but most studies have indicated that the existing medications do not modify the long-term decline of the lung function [Donald et al. 2008; Burge et al. 2000; Pauwels et al. 1999]. Clinical symptoms related to COPD are cough and sputum production but progressive and permanent dyspnoea is always described by patients as the major contributor to disease-related disability. Thus, most of the present management of COPD and especially pharmacological management is devoted to relief of this important complex and subjective symptom.
Bronchodilator medications are central to the symptomatic management of COPD [Vathenen et al. 1988; Higgins et al. 1991]. They may increase the forced expiratory volume in one second (FEV1) and other spirometric variables, by improving lung mechanics. Indeed, in addition their effect on smooth muscle cells, these drugs have been shown to facilitate emptying of the lungs, reducing hyperinflation at rest and during exercise [O’Donnell et al. 2006; O’Donnell et al. 2004]. These effects are perhaps more important for the COPD daily strategy for a given patient than the net increase in classical spirometric parameters.
Three subtypes of muscarinic receptors are expressed in the human airway: M1, M2 and M3 receptors [Belmonte, 2005] (Figure 1). M1 receptors are localized to parasympathetic ganglia in the airways, facilitating ganglionic neurotransmission [Barnes, 2004]. M2 receptors localized in cholinergic nerve endings act as negative feedback of acetylcholine release from the nerve [Roffel et al. 2001; Minette and Barnes, 1988]. Finally, the M3 receptors are the primary subtype responsible for bronchial epithelial cells activation and tracheal smooth muscle contraction [Roffel et al. 1990]. Acetylcholine is involved in airway smooth muscle action and narrowing, and cholinergic tone appears to be the major reversible component of airway obstruction in COPD [Gross, 1988; Gross and Skorodin, 1984]. Participation of mucus secretion and oedema of the airway wall are suspected, but less well documented.
Muscarinic receptors in the airways.
Inhaled anticholinergics belong to an effective class of bronchodilators in the management of symptomatic patients with COPD. The most important effect of anticholinergic drugs appears to be driven by the blockage of the acetylcholine effect on M3 receptors [GOLD, 2009]. Ipratropium can also block M2 receptors while tiotropium has a pharmacokinetic selectivity for M1 receptors [GOLD, 2009]. These compounds have potential systemic anticholinergic effects including dry mouth (reported in 11.6% of patients treated with tiotropium bromide versus 3.5% of patients treated with placebo), glaucoma, constipation, increased heart rate, and urinary retention [Food and Drug Administration, 2006; Kesten et al. 2006].
Aclidinium bromide
Aclidinium bromide, 3 R-(2-hydroxy-2,2-di-thiophen-2-yl-acetoxy)-1-(3-phenoxy-propyl)-1-azonia-bicyclo[2.2.2] octane bromide (Figure 2), previously known as LAS34273, is a novel inhaled muscarinic antagonist [Prat et al. 2009] currently being studied in different phase III clinical trials as a maintenance treatment of COPD.
Chemical structure of aclidinium bromide. (Reproduced with permission from Prat et al. [2009].).
In contrast to other currently available antimuscarinics including tiotropium, aclidinium has been shown to undergo rapid hydrolysis in human plasma, resulting in very low and transient systemic exposure, suggesting a reduced potential for class-related systemic side effects [Gavaldà et al. 2009; Gavaldà et al. 2008].
Interaction with the human muscarinic receptor subtypes
Aclidinium is mainly an M2 and M3 receptor antagonist. The most important interactions with human muscarinic receptor subtypes in COPD are those involving M2 and M3 receptors. The M2 receptor is interesting beyond its potential role in the efficacy of antimuscarinics, because cardiac M2 receptor inhibition is known to induce tachycardia, which is potentially the most severe side effect associated with systemic antimuscarinic agents. This side effect is potentially of crucial importance in COPD as cardiovascular comorbid conditions are frequently associated in these patients. The M3 receptor is the key receptor subtype through which the therapeutically relevant muscle relaxant and bronchodilatory effects of the antimuscarinic agents are mediated [Eglen, 2005].
Saturation studies with [3H]-aclidinium, [3H]-ipratropium and [3H]-tiotropium at membranes expressing human M2 and M3 receptors.

K d, equilibrium dissociation constant; B max, binding maximum.
The association rate of aclidinium with the human M3 receptor subtype was similar to that of ipratropium and 2.6 times faster than that of tiotropium. The K
on of the three antagonists for the human M2 receptor subtype was too fast to be reliably measured under the experimental conditions used. Aclidinium and tiotropium showed slow dissociation from M3 receptors (Figure 3), with residence half-lives of approximately 29 and 62 h, respectively. Conversely, dissociation of ipratropium from the same receptor was much faster, resulting in a residence half-life approximately 60- to 130-fold shorter than that of aclidinium and tiotropium, respectively. The aclidinium half-life for the M2 receptor was 3.22 times shorter than that obtained for tiotropium.
Dissociation of aclidinium, ipratropium and tiotropium from human M2 and M3 receptors. (A) Dissociation profile of radiolabeled compounds from human M2 receptor: dissociation from 0 to 3000 min (1) and dissociation from 0 to 60 min (2). (B) Dissociation profile of radiolabeled compounds from human M3 receptor: dissociation from 0 to 3000 min (1) and dissociation from 0 to 120 min (2). (Reproduced with permission from Gavaldà et al. [2009].).
Plasma inactivation and pharmacological activity of aclidinium main metabolites
Pharmacokinetic studies of inhaled aclidinium up to 6000 µg have shown rapid elimination from systemic circulation (t
1/2 < 1.5 h), consistent with a rapid in vivo plasma hydrolysis of aclidinium [Sentellas et al. 2010; Jansat et al. 2009b], suggesting that enzymatic hydrolysis may be a key factor in the metabolism of this compound [Sentellas et al. 2010]. Alberti et al. (2010) have identified that human butyrylcholinesterase is the most important enzyme involved in the enzymatic hydrolysis of aclidinium and that this process takes place mainly in plasma. Biological and chemical hydrolysis of the ester bond of aclidinium in vitro results in two major compounds, the alcohol (LAS34823 [3(R)-hydroxy-1-(3-phenoxy-propyl)-1-azonia-bicyclo[2.2.2]octane bromide]) and the carboxylic acid (LAS34850 [dithienyl-glycolic acid, sodium salt]) metabolites (1, 28) (Figure 4).
Hydrolytic cleavage of the ester moiety of aclidinium bromide into its alcohol (LAS34823) and carboxylic acid (LAS34850) derivatives. (Reproduced with permission from Alberti et al. [2010].).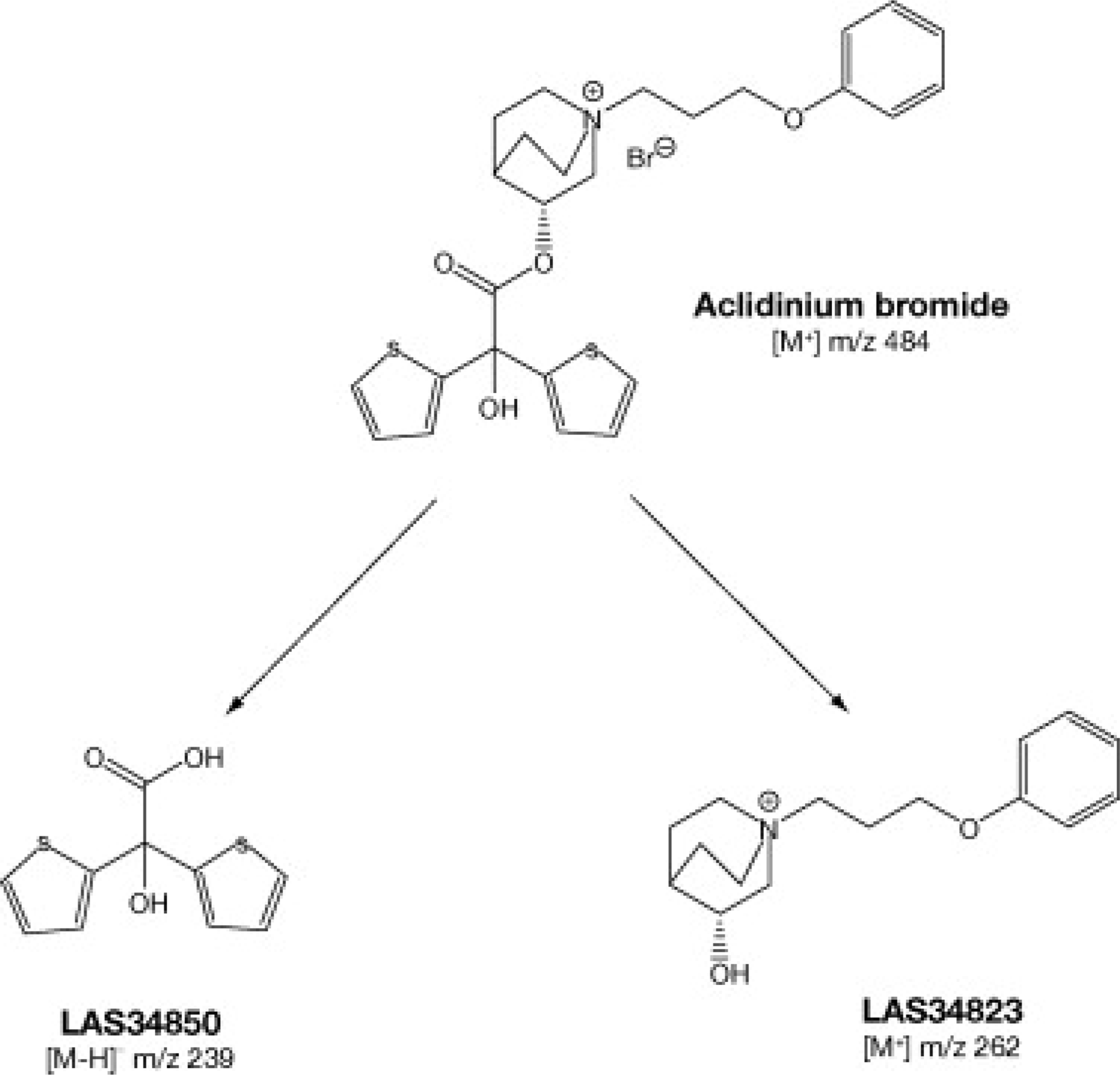
Both metabolites have also been detected in human plasma after an inhaled dose of aclidinium (400 µg and 800 µg). In the latest studies, improving the lower limit of detection allowed the detection of aclidinium and its metabolites even after a unique dose as low as 200 µg. The acid metabolite remained detectable in the plasma 100-fold higher than aclidinium but the alcohol metabolite remained detectable two- to five-fold higher [Jansat et al. 2009a, 2009b]. The relatively high plasma concentration of the acid metabolite is mainly due to differences in oral absorption, which is higher with the acid metabolite than with aclidinium and the alcohol metabolite [Jansat et al. 2009a, 2009b]. In addition, other minor oxidative metabolites of aclidinium have been observed in vitro and in vivo in different animal species and humans.
Affinity of aclidinium and its main metabolites LAS34823 and LAS34850 for human M1, M2, M3, M4 and M5 receptors.

The effect of LAS34823 and LAS34850 on the acetylcholine-induced bronchoconstriction model in rodents was also assessed in comparison with aclidinium bromide. The two main aclidinium metabolites did not show any relevant bronchodilatory activity in vivo. In contrast, a potent bronchodilatory activity was observed after aclidinium inhalation, in concordance with results reported previously [Sentellas et al. 2010; Gavaldà et al. 2009]. These data demonstrate that the activity observed in vivo is exclusively due to aclidinium bromide without any contribution from its hydrolysis products [Sentellas et al. 2010].
Clinical trials
Phase I
The pharmacokinetics and pharmacodynamics of aclidinium have been investigated in a phase I study (doses of 50, 300, and 600 µg) [Donald et al. 2008; Schelfhout et al. 2007]. The results from this study showed that, in healthy subjects, aclidinium produced significant, long-lasting (≥24 h) protection against methacholine-induced bronchoconstriction [Schelfhout et al. 2007].
Phase IIa
In an initial study [Joos et al. 2010], seventeen patients, with a diagnosis of moderate to severe COPD, were randomly assigned to one of four treatment sequences (100, 300, 900 µg or placebo) once daily. Aclidinium bromide (100, 300 and 900 µg) significantly improved pulmonary function tests as assessed by FEV1 (Figure 5) and forced vital capacity (FVC) (Figure 6). The bronchodilatory effect was sustained for at least 24 h. The maximal effect of aclidinium bromide was observed at 2 h postdose, with a corresponding mean peak increase in FEV1 of 17.2%, 23.3% and 21.4% for aclidinium bromide 100, 300 and 900 µg doses, respectively. This maximal effect of aclidinium bromide was similar to that reported in the baseline ipratropium reversibility test (21.1% after 30 min).
(A) Mean (SE) change from baseline in forced expiratory volume in 1 s (FEV1) over 4 h postadministration. (B) Mean (SE) change from baseline in FEV1 over 32 h postadministration. (Reproduced with permission from Joos et al. [2010].). Mean (SE) change from baseline in forced vital capacity (FVC) over 32 h postadministration. (Reproduced with permission from Joos et al. [2010].).

A total of 28 treatment-emergent adverse advents (AEs) were reported in 11 patients during this initial study. The majority of AEs were mild (64%) or moderate (36%) in intensity and three quarters (21/28) were considered to be unrelated to the study drug. One out of four was considered possibly related to study drug (headache n = 6, increased sweating n = 1).
Phase IIb
In a larger study [Chanez et al. 2010], 464 patients were randomized with a diagnosis of stable moderate to severe COPD according to guidelines. A total of 441 patients completed this double-blinded, placebo-controlled study. The patients were randomized to one of seven treatments: aclidinium 25, 50, 100, 200 or 400 µg; matching placebo; or tiotropium 18 µg (open label). All treatments were taken once daily. Efficacy spirometry measurements were taken at 0.5, 1, 2, 3, 4, 5 and 6 h after the first and last dose (days 1 and 29, respectively) in addition to 22, 23 and 24 h after the first dose and just before the last dose. This study showed that aclidinium provided a sustained 24-h bronchodilation effect that was significantly superior to placebo. The effect was sustained over the 4 weeks of the study. The aclidinium doses selected in this study ranged from an expected noneffective dose (25 µg) to a known safe and effective dose (400 µg). The trough FEV1 results showed a maximal effect with aclidinium 200 µg daily. The minimum effective dose, defined as the lowest dose of aclidinium which significantly improved FEV1 over placebo on day 29, was 100 µg in this COPD population. Spirometric results observed for aclidinium 200 µg and 400 µg administered once daily were similar to those observed with tiotropium 18 µg over the 4 weeks of the study (Figure 7).
Change from baseline in FEV1 0–6 h postadministration (intent-to-treat population). (A) Day 1 (B) Day 29. (A) Statistically significant increases from baseline versus placebo at all time points for aclidinium 100 µg (p = 0.014), 200 µg (p < 0.001) and 400 µg (p < 0.001) and tiotropium 18 µg (p = 0.02). (B) Statistically significant increases over placebo at all time points for aclidinium 100, 200 and 400 µg, as well as for tiotropium 18 µg (p < 0.001 in all cases except for the aclidinium 100 µg dose: p = 0.006). (Reproduced with permission from Chanez et al. [2010].).
During the 4-week treatment period, the health-related quality of life, measured using the St George's Respiratory Questionnaire (SGRQ) overall score, improved from baseline with all doses of aclidinium. The percentage of patients with a clinically meaningful improvement in SGRQ total score of ≥4 points from baseline ranged from 52.6% in the 400 µg group to 64.4% in the 100 µg group. The improvements observed in the SGRQ were mainly related to changes in the symptoms and impacts components.
There was a trend toward an improvement in dyspnoea index measured by an increase in transition dyspnea index (TDI) total score compared with placebo for all doses of aclidinium. TDI component scores: (functional impairment, magnitude of task, and transition focal component) were statistically significantly improved by aclidinium 100 µg and 400 µg when compared with placebo; however, there was no significant difference for any treatment group in the magnitude of effort component score in this only 4-week trial.
Aclidinium was well tolerated; most AEs were mild to moderate and considered by the investigators to be unrelated to treatment. The incidence of specific AEs was low across all active treatment groups and comparable with placebo, except for headache which occurred with an incidence of >5% in the aclidinium 50 µg and 100 µg groups (6.2% and 10.0%, respectively), and dry mouth, which occurred with an incidence of 6.2% in the tiotropium group. There was no relationship between the dose of aclidinium and the number of AEs [Chanez et al. 2010].
Summary
Aclidinium is a potent and selective muscarinic antagonist, which interacts rapidly with muscarinic receptors, and shows subnanomolar affinity for the five human muscarinic receptors (M1–M5); its association rate for the M3 receptor is similar to that of ipratropium and 2.6 times faster than that of tiotropium. Aclidinium dissociates slightly faster from M2 and M3 receptors than tiotropium but much more slowly than ipratropium. A potent bronchodilatory activity has been observed after inhaled administration of aclidinium. Aclidinium undergoes rapid hydrolysis in the plasma into two major compounds, the alcohol (LAS34823) and the carboxylic acid (LAS34850) metabolites, resulting in low and transient systemic exposure to the active drug. The two major metabolites show no significant affinity for human muscarinic receptors. Clinical trials have provided evidence of sustained bronchodilation similar to that observed with tiotropium. Safety results confirm the positive safety profile of aclidinium, particularly in terms of a very low propensity to cause anticholinergic AEs. Aclidinium is now moving to phase III clinical development for COPD.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
None declared.