
Abstract
The soluble form of the membrane hemoglobin scavenger receptor CD163 (sCD163), released by shedding, is a strong marker for macrophage activation. Serum sCD163 levels rise in several acute inflammatory states and some fibrosing diseases. Monocyte-derived macrophages (MoDM) differentiated by macrophage colony-stimulating factor (M-MoDM) contribute to the pathophysiology of idiopathic pulmonary fibrosis (IPF), an irreversible and rapidly fatal interstitial lung disease. Since M-MoDM express high membrane CD163 levels, we thus postulated that sCD163 could be a relevant biomarker for macrophage activation in IPF. We found that M-MoDM constitutively released higher amounts of sCD163 (49.5 ± 24.5 ng/ml) than monocytes (0.45 ± 0.32 ng/ml) or MoDM differentiated with granulocyte macrophage-stimulating factor (2.24 ± 0.98 ng/ml). The basal production of sCD163 by M-MoDM was increased following stimulation with lipopolysaccharide (123.4 ± 54.9 ng/ml) or ATP (168.9 ± 41.8 ng/ml). The sCD163 release was controlled by metalloproteases but not through ADAM17 activation. Moreover, CD163-positive macrophages and sCD163 were detected in pulmonary tissues and alveolar fluids of Caucasian patients with IPF, respectively. IPF alveolar macrophages constitutively secreted sCD163 amounts (67.6 ± 44.6 ng/µg RNA) which were significantly higher than those released by alveolar macrophages isolated from controls (19.2 ± 7.6 ng/µg RNA) or patients with other interstitial lung disease (31.5 ± 16.6 ng/µg RNA). However, the concentrations of sCD163 in blood serum collected from 155 patients with IPF did not correlate with the severity of their disease. In conclusion, our results show that M-MoDM constituted a pertinent model to study the regulation of sCD163 production. Yet, serum sCD163 values could not provide a prognostic biomarker for IPF in our cohort.
Keywords
Introduction
CD163, a scavenger receptor for haptoglobin-hemoglobin complexes, is selectively expressed on the membrane of monocytes and macrophages. 1 The shedding of membrane CD163 by metalloproteases such as ADAM17 can release a soluble form called sCD163. Although its physiological role remains unknown, sCD163 is recognized as a strong marker for macrophage activation.1–3 Several clinical studies have indeed reported that blood sCD163 levels are increased in patients with acute inflammatory diseases such as sepsis and acute hepatitis.1,4 In addition, high serum sCD163 levels could be associated with the severity of chronic fibrotic diseases. Increased serum CD163 concentrations constitute a relevant biological marker for aggravation of HBV and HCV-related liver fibrosis3,5 and interstitial lung disease (ILD) associated with inflammatory myositis. 6
Idiopathic pulmonary fibrosis (IPF) is an irreversible and rapidly fatal diffuse ILD. 7 Its pathophysiology is complex and still poorly understood. The current hypothesis holds that repeated damages to the alveolar epithelium in predisposed subjects lead to the release of cytokines activating fibroblasts and inducing their differentiation into myofibroblasts. 7 The myofibroblasts synthesize a dense extracellular matrix, in a self-perpetuated loop, that impairs pulmonary architecture and promotes lung tissue stiffness. These events finally lead to an irreversible decline in the pulmonary functional capacity that characterize patients with IPF. 8
In addition, the innate immune system plays a central role in IPF by promoting the profibrotic functions of pulmonary fibroblasts from the earliest stages of the disease.9,10 Specifically, abnormal activation of macrophages participates to the remodelling of the extracellular matrix and contributes to the recruitment of other immune cells. In response to repeated alveolar tissue aggression, circulating monocytes can first be recruited to the lungs by the CC chemokine ligand 2 (CCL2) and then differentiated by macrophage colony-stimulating factor (M-CSF) into interstitial or alveolar macrophages.11–13 Depletion of these monocyte-derived macrophages (MoDM), or blockade of the M-CSF receptor, reduces lung fibrosis in several murine models.14–16 There is also some evidence for the infiltration of monocytes/macrophages into the pulmonary tissues of patients with IPF and for the increase of M-CSF and CCL-2 concentrations in the fluids of bronchoalveolar lavages (BALFs), 13 which suggest a role for MoDM in the pathophysiology of IPF. We recently demonstrated that human MoDM differentiated in the presence of M-CSF, called M-MoDM, are characterized by a high membrane CD163 expression when compared to that measured in MoDM differentiated by granulocyte macrophage-colony stimulating factor (GM-CSF). 17 However, we did not investigate the production of sCD163 by M-MoDM. Such CD163-positive macrophages are detected in BALFs and they are present in the pulmonary tissues of Asian patients with IPF.15,18,19 In particular, according to Nouno et al., 18 the number of CD163-positive macrophages present in the lungs is increased in patients with IPF and is negatively associated with their survival. Moreover, two studies have recently reported that the serum levels of sCD163 are higher in Asian patients with IPF than in control healthy subjects,20,21 thus suggesting that sCD163 could be a relevant biomarker for macrophage activation in IPF.
In this context, the aims of the present study were to characterize the sCD163 expression in human M-MoDM and alveolar macrophages and then to evaluate the prognostic value of serum sCD163 concentrations in Caucasian patients suffering from IPF.
Materials and methods
Chemicals and reagents
Premium grade M-CSF and GM-CSF were purchased from Miltenyi Biotec (Paris, France) and Berlex laboratories Inc. (Bayer, France), respectively. Lipopolysaccharide (LPS) (Escherichia coli O55:B5), adenosine 5’-triphosphate (ATP), uridine 5′-triphosphate (UTP), the metalloproteinase inhibitor TAPI-0, the P2X7 receptor inhibitor A-804598 and dexamethasone were from Sigma-Aldrich (Saint-Quentin Fallavier, France). The human recombinant High Mobility Group Box 1 (HMGB1) was from R&D System (Biotechne, Lille, France). Interferon-γ (IFN-γ), interleukin (IL)-4, IL-10 and IL-13 were from PeproTech (Neuilly-sur-Seine, France). Primary antibodies (Ab) against human ADAM17 and GAPDH were from Santa Cruz Biotechnology (Heidelberg, Germany) and Cell Signaling Technology (Ozyme, Montigny-le-Bretonneux, France), respectively. Si-RNAs: ON-TARGETplus Control pool Nontargeting Pool (D-001810-10-05, Si-CTR) and ON-TARGETplus Human ADAM17 siRNA-SMARTpool (L-003453-00-0005, Si-ADAM17) were purchased from Dharmacon (Horizon Discovery, UK).
Patient selection
IPF was diagnosed according to the 2011 and updated 2018 ATS/ERS/JRS/ALAT criteria, which include histopathological features of usual interstitial pneumonia. 8 Non-stable patients with a respiratory infection or exacerbation in the eight weeks preceding the diagnostic consultation were excluded. Data were collected in all the patients at the diagnosis of IPF from 2014 to 2020. The research protocols described in the following sections were conducted under French legal guidelines and were approved by the local institutional ethics committee (Comité de protection des personnes Ouest V, CHU Rennes, n°2014-A00268-39; Comité d’éthique, avis n°16.123; Comité d’éthique, avis n°19.09). All subjects signed a written informed consent form for their tissue (blood monocyte, surgical lung biopsies, BALF and serum) to be used for research.
Cell cultures
Buffy coats provided by the Etablissement Français du Sang (Rennes, France) and blood samples were collected from twenty six healthy donors and from eleven patients with IPF, respectively. Clinical and pulmonary functional data of the eleven patients with IPF have previously been published in the Table S1 “Blood monocytes” in our previous article.
22
Briefly, peripheral blood mononuclear cells from healthy and IPF donors were isolated by Ficoll gradient centrifugation and monocytes were next selected by a 2 h adhesion step.
23
Monocytes were either left untreated for 24 h in GlutaMAX RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum, 2 mM
Flow cytometry
The membrane expressions of CD163, CD71, CD206 and CD14 were analyzed by direct immunofluorescence using flow cytometry. After washing, detached cells were stained with Fixable Viability Stain 450 (BD Biosciences, Le Pont de Claix, France) for 10 min at room temperature to measure viability. Non-specific Ab binding sites on cells were blocked by incubating them in PBS containing 2% FBS and Fc block (Miltenyi Biotec SAS, Paris, France) for 10 min at room temperature. Cells were next incubated with a specific Ab or its isotypic control for 20 min at 4 °C, washed in PBS and collected by centrifugation (2500 rpm for 5 min). Ab binding was analyzed in an LSR II cytometer and FACSDiva software. The panel used to characterize monocytes and macrophages was: FITC anti-CD14 (clone REA599, Miltenyi Biotec), PE anti-CD206 (Clone 19.2, BD Biosciences), APC anti-CD71 (clone REA902, Miltenyi Biotec) and BV421 anti-CD163 (Clone GHI/61, BD Biosciences). Results are expressed as the ratio of median fluorescence intensity (MFI) calculated as: MFI (test Ab) / MFI (isotype control Ab).
Cytokine quantification
The concentrations of sCD163 and TNF-α were measured in cell culture media by ELISA (Duoset ELISA development system kits, R&D Systems Europe, Lille, France) according to the manufacturer's instructions. sCD163 concentrations were also quantified in blood serum samples taken from 155 patients when they were diagnosed with IPF. Clinical parameters and pulmonary functional data of the 155 subjects are presented on the Table 1. Serum samples were obtained from the processing of biological samples through the Centre de Ressources Biologiques (CRB) Santé of Rennes BB-0033-00056. Serum sample were stocked at −80°C until the measurement of sCD163 concentrations. The sensitivity and range of the sCD163 assay were 0.13 ng/ml and 1.6–100 ng/ml, respectively.
Multivariate survival analysis (Cox model) and characteristics of the patients with IPF (n = 155).
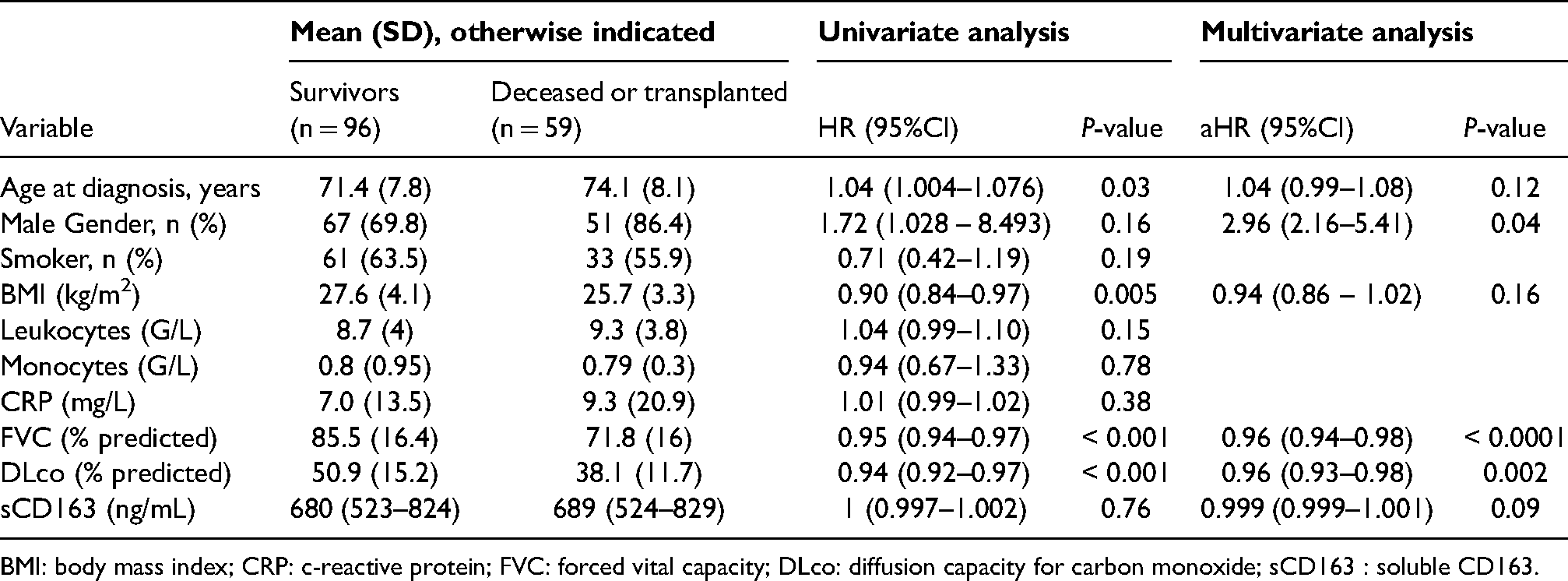
BMI: body mass index; CRP: c-reactive protein; FVC: forced vital capacity; DLco: diffusion capacity for carbon monoxide; sCD163 : soluble CD163.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR
)
Total RNA was extracted and reverse transcribed as previously described. 23 Quantitative PCR was performed using the SYBR Green methodology on a CFX384 real-time PCR system (BioRad, Marnes-la-Coquette, France). The primers were provided by Sigma-Aldrich (Kiqstart primers). The specificity of amplified genes was checked at the end of PCR using the comparative cycle threshold method (CFX Manager™ Software). These mean Cq values were used to normalize the target mRNA concentrations to those of the 18S ribosomal protein by the 2( − ΔΔCq) method.
Western blotting
Whole cell lysates were prepared as previously described. 22 Briefly, cells were lysed with RIPA buffer containing a protease inhibitor cocktail (Roche Diagnostic, Meylan, France) and phosphatase inhibitor cocktails 2/3 (Sigma-Aldrich, Saint-Quentin Fallavier, France) and then centrifuged. Lysates were diluted in loading buffer, heated at 95°C, loaded on a 4% stacking gel and separated on a 10% gel by SDS electrophoresis. The proteins were then transferred to nitrocellulose membranes by electroblotting overnight (30 V at 4°C). After blocking, the membranes were then hybridized overnight at 4°C with appropriate primary Abs and incubated with HRP-conjugated secondary Abs before analysis by chemiluminescence on a ChemiDoc XRS + System and Image Lab software (Bio-Rad, Marnes-la-Coquette, France).
Immunofluorescence (IF)
IF analysis were performed on lung biopsy samples obtained from six patients with IPF. The clinical and pulmonary functional data of the six subjects have already been published in the Table S1 (see “Lung biopsies”) in our previous article. 22 Whole-slide sections (4 µm) were cut from lung tissue blocks with a microtome, transferred to charged slides, and costained for CD68, CD206 and CD163 (U DISCOVERY 4 plex IF; Roche Diagnostic, Meylan, France) using the following Abs (at similar dilutions): CD68 (M0876, clone PG-M1; Dako), CD206 (ab64693, Abcam), CD163 (NCL-CD163, clone 10D6; Novocastra). Proteins were visualized by incubating sections with unmodified primary Ab, the corresponding HRP-conjugated secondary Ab and then by producing the HRP enzyme-mediated deposition of the tyramide-fluorophore that covalently binds to the tissue at the site of the reaction. 22 The primary Ab and secondary Ab-HRP complexes were finally heat-inactivated. CD68 was detected with tetramethylrhodamine (TRITC), CD206 with fluorescein isothiocyanate (FITC), and CD163 with cyanine 5 (Cya5). These sequential reactions were repeated three times. Stained sections were mounted in fluoromount (Enzo Life Sciences, Farmingdale, NY, USA) and slides were scanned using a Hamamatsu scanner (Hamamatsu, Iwata City, Japan). The recorded images were analyzed using the Hamamatsu NDPview2 software. All manipulations were performed at the Rennes H2P2 Histopathology platform (SFR UMS CNRS 3480 - INSERM 018).
Preparation of BALFs
BALF was obtained from 6 control patients, 25 patients with non-IPF ILD and 9 patients with IPF. Control patients were suffering from alveolar hemorrhage (n = 2), aspiration pneumonia (n = 1), lung cancer (n = 2), or pleuritis (n = 1). Patient with non-IPF ILD suffered from unclassifiable ILD (n = 7), sarcoidosis (n = 5), connective tissue disease-associated ILD (n = 3), hypersensitivity pneumonitis (n = 2), smoking-related interstitial fibrosis (n = 2), drug-induced ILD (n = 2), ANCA-associated ILD (anti-myeloperoxidase, n = 1), pleuroparenchymal fibro-elastosis (n = 1), interstitial pneumonia with auto-immune features (n = 1), or silicosis (n = 1). The main characteristics of these patients are presented in the Table S1. BALFs was obtained as previously described. 22 The average volume obtained for experimental research was approximately 10 ml. The resulting BALFs were then centrifuged, washed once with 5 ml PBS, and suspended in GlutaMAX RPMI 1640 medium (Thermo Fisher Scientific, France) containing 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 20 IU/ml penicillin and 20 μg/ml streptomycin for 3 h. The adherent cells, i.e. alveolar macrophages, were washed and cultured in complete medium in the presence or absence of 20 ng/ml LPS for 24 h. At the end of the treatment, the supernatants were collected and frozen at −20°C. Cells were harvested to extract total RNA as described above.
Statistics
For in vitro experiments, significant differences were assessed using a two-way Student's t-test or a one-way ANOVA followed by the multirange Dunnett's t-test for multiple comparisons. Correlations between the sCD163 blood concentrations and clinical parameters were determined using Spearman's rank correlation coefficient. Differences were considered to be significant when P < 0.05. For clinical studies, continuous variables were expressed as mean and standard deviation (SD), and qualitative variables were expressed as n (%). Fisher's exact test was used for categorical variables and Mann-Whitney test or t test for continuous variables when appropriate. We performed univariate Cox regression analyses to identify risk factors associated with mortality or lung transplantation. Variables with a P-value <0.20 were included in multivariate Cox regression models. Variables with missing values > 10% were not included in the multivariate analysis. Then, we built an adjusted Cox proportional hazards regression model. Stepwise backward variable elimination using Akaike's information criterion was performed to construct the final model. Schoenfeld residuals were used to test the proportional hazard assumption. Results were expressed as hazard ratios (HRs) and adjusted HRs (aHR) with 95% confidence intervals (CIs). Statistical analysis was carried out by R-Studio 2020: Integrated Development for R. (R-Studio, Boston, MA).
Results
Membrane and sCD163 expressions were higher in M-MoDM
In order to study the membrane and soluble expression of CD163 in human monocytes, M-MoDM and GM-MoDM, we first analyzed the expression of three well-known markers of these mononuclear cells. Monocytes expressed high CD14 levels (MFI: 15.03 ± 11.06) and low CD71 (MFI: 2.3 ± 0.7) and CD206 (MFI: 3.1 ± 0.9) levels (Figure 1a). In contrast, CD14 levels were specifically decreased in GM-MoDM (MFI: 2.5 ± 0.5) whereas CD71 and CD206 expression were significantly increased in both GM-MoDM and M-MoDM. The membrane CD163 expressions (MFI) in monocytes, GM-MoDM, and M-MoDM were 3.5 ± 1.1, 1.7 ± 0.6 and 21.4 ± 5.9, respectively. Most M-MoDM (90.5 + 5.1%) were positive for CD163. Stimulation with LPS significantly decreased the percentage and the intensity of CD163 expression in both monocytes and M-MoDM (Figure 1b). LPS did not modulate CD163 RNA levels but it significantly increased TNF-α mRNA levels in monocytes, GM- and M-MoDM (Figure 1c). Thus, the LPS-dependent decrease in membrane CD163 expression in monocytes and M-MoDM (Figure 1b) was unlikely to result from the repression of CD163 gene expression. We next determined if this modulation of membrane CD163 expression was associated with the release of sCD163 from M-MoDM. Figure 1d shows that the basal concentrations of sCD163 was 0.4 ± 0.3 ng/ml and 2.3 ± 0.9 ng/ml in the medium of monocytes and GM-MoDM cultures, respectively. In contrast, M-MoDM released higher amounts of sCD163, constitutively (49.5 ± 24.5 ng/ml) and after LPS stimulation (123.4 ± 54.9 ng/ml).
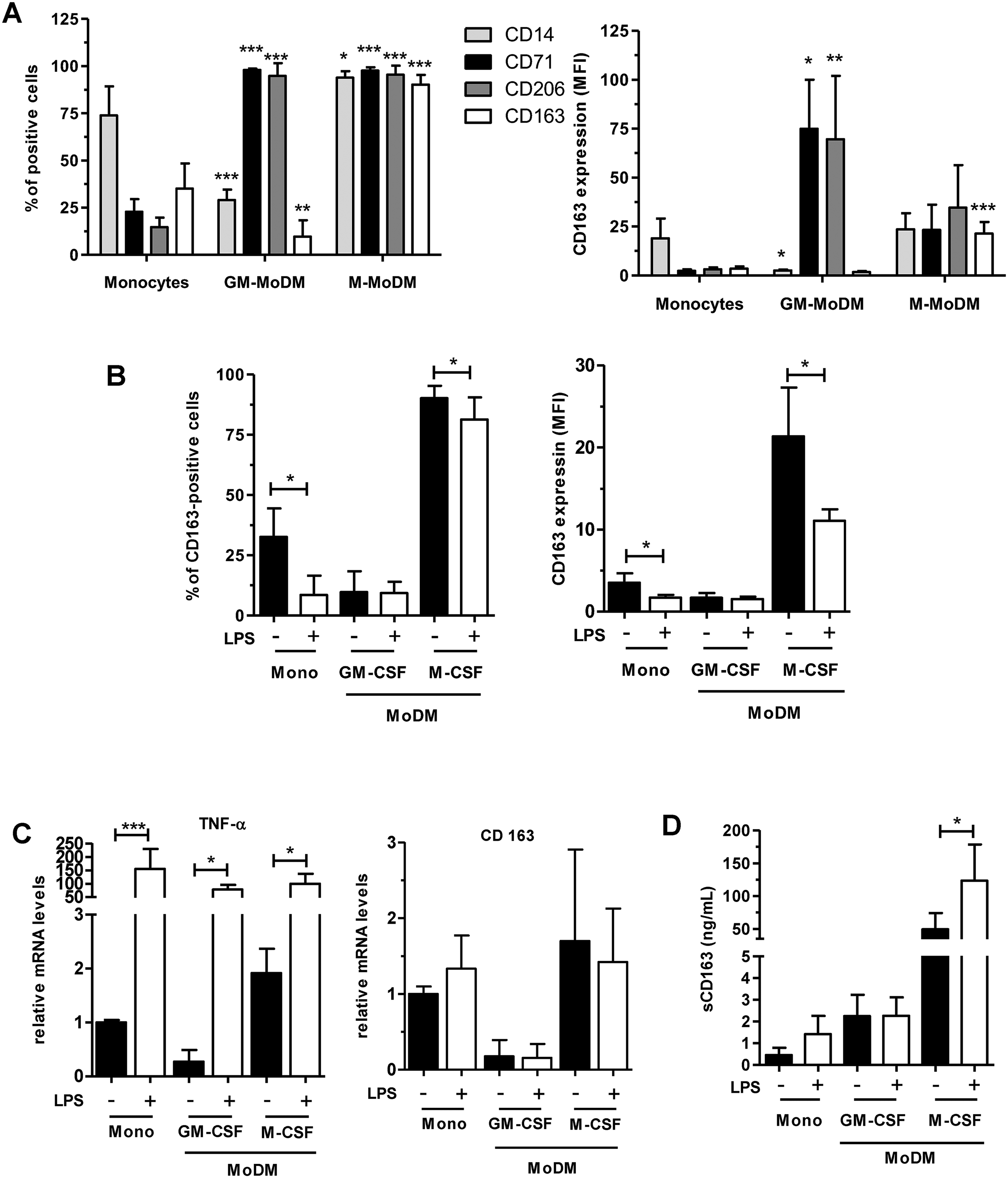
Membrane and sCD163 expressions were increased in MoDM differentiated with M-CSF. Once isolated, monocytes were left untreated for 16 h or differentiated with GM-CSF (GM-MoDM) or M-CSF (M-MoDM) for 6 days. Then, monocytes (Mono) and MoDM were untreated or stimulated with 20 ng/ml LPS for 1 h. In (a, b), cells were analyzed by flow cytometry to measure membrane expression of the CD14, CD71, CD206 (A) and CD163 (A, B) markers. In (c), Relative TNF-α and CD163 mRNA levels were determined by quantitative RT-PCR and normalized to endogenous ribosomal 18S mRNA levels. Data are expressed relative to mRNA levels found in untreated monocytes, arbitrarily set at 1. In (d), culture supernatants were collected to measure by ELISA the concentrations of sCD163 secreted in the culture media. The results are expressed as the means ± SD of 4 (a, b, d) and 3 (c) independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus “Monocytes” in (A).
LPS rapidly stimulated the release of sCD163 from M-MoDM
We next determined the kinetics of sCD163 release from M-MoDM. The membrane CD163 expression decreased very rapidly in response to LPS stimulation (Figure 2a) and was associated with a parallel increase of sCD163 levels in culture medium (Figure 2b). sCD163 concentrations in the culture medium of LPS-stimulated M-MoDM increased up to 1 h and then remained stable for 24 h (Figure 2c).
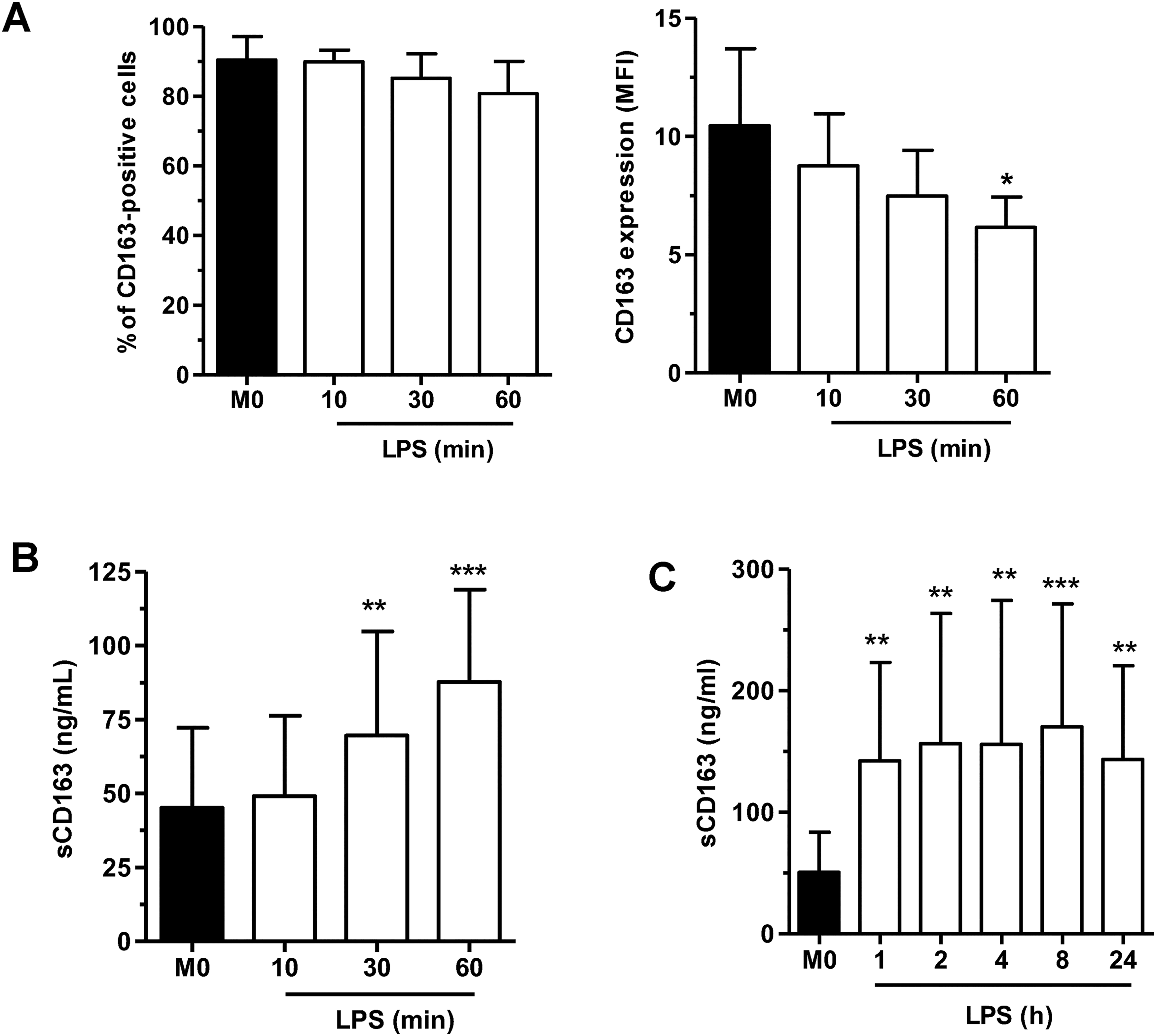
LPS rapidly modulated the expression of membrane and sCD163. M-MoDM were left untreated (M0) or stimulated with 20 ng/ml LPS for the indicated time intervals. Membrane (a) and soluble (b, c) CD163 expressions were quantified by flow cytometry and ELISA, respectively. The results are expressed as the means ± SD of 5 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus “M0”.
Metalloproteases, but not ADAM17, mediated the release of sCD163
We then determined if metalloproteases, such as the disintegrin and metalloproteinase ADAM17, could regulate the secretion of sCD163 from M-MoDM. Pre-treatment with the pan-metalloprotease inhibitor TAPI-0 had no impact on the basal membrane CD163 expression and sCD163 release (Figure 3a and 3b). However, it significantly reduced the LPS-dependent decrease of membrane CD163 levels, i.e. MFI: 21.2 ± 5.1 (basal), 10.2 ± 5.1 (LPS), 15.9 ± 9.5 (TAPI-0 + LPS) (Figure 3a, right panel) and the parallel release of sCD163, i.e. 94.1 ± 19.8 ng/ml (basal), 186.8 ± 26.7 ng/ml (LPS) and 82.05 ± 10.6 ng/ml (TAPI-0 + LPS) from LPS-stimulated M-MoDM (Figure 3b). Next, using the mRNA silencing technology, we totally inhibited ADAM17 protein expression in M-MoDM transfected with Si ADAM17 for 24 h (Figure 3c). Stimulation for 1 h with LPS significantly decreased membrane CD163 expression in control cells transfected with Si Ctr (Figure 3d). Genetic silencing of ADAM17 did not modulate the basal CD163 expression and did not prevent its decrease in LPS-stimulated cells. Similarly, the repression of ADAM17 expression affected neither the basal sCD163 secretion nor its increase by LPS (Figure 3e, left panel). In contrast, silencing of ADAM17 significantly decreased the LPS-dependent secretion of TNFα (Figure 3e, right panel).
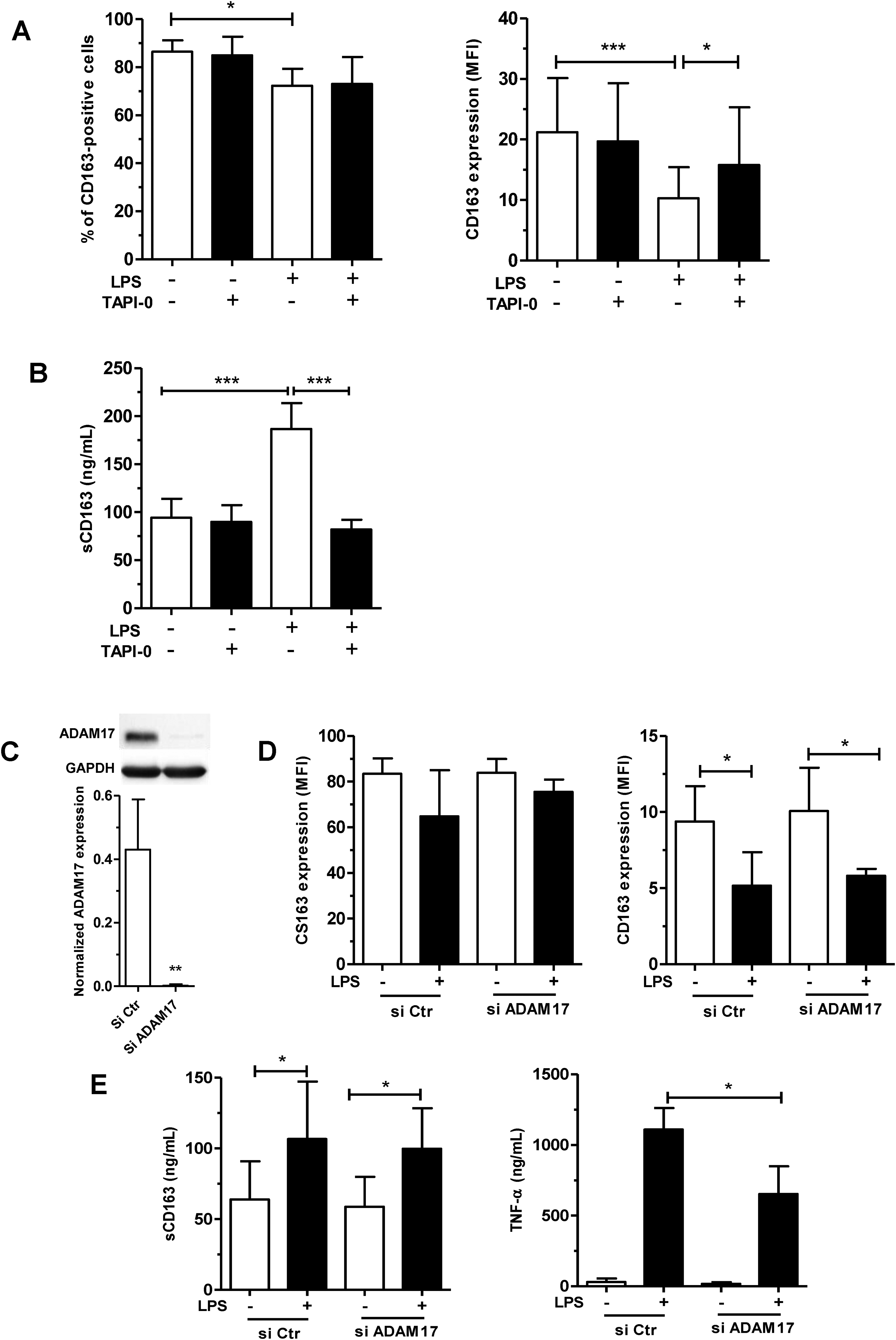
Metalloproteases but not ADAM17, controlled the release of sCD163 from M-MoDM. Macrophages were pre-treated with 10 µM TAPI-0 for 1 h (a, b) or transfected with ADAM17 SiRNA for 24 h (c–e) and then stimulated with 20 ng/ml LPS for 1 h. Membrane (a, d) and soluble (b, e) CD163 expressions were quantified by flow cytometry and ELISA, respectively. TNF-α release was quantified by ELISA (e). Downregulation of ADAM17 protein expression was detected by Western blot in (c). Relative ADAM17 levels were normalized to GAPDH levels quantified by densitometry. The results are expressed as the means ± SD of 4 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
ATP, but not UTP or HMGB1, stimulated the release of sCD63
We also investigated if, besides LPS, ATP could induce the shedding of CD163 in M-MoDM. Figure 4a demonstrates that 3 mM ATP significantly decreased both the percentage of CD163-positive cells and the intensity of membrane CD163 expression, i.e. MFI: 17.9 ± 3.7 (basal) and 4.7 ± 3.1 (ATP), (Figure 4a) whereas it strongly increased the secretion of sCD163, i.e. 88 ± 43.4 ng/ml (basal) and 168.9 ± 41.8 (ATP) (Figure 4b). The selective purinergic P2X7 receptor inhibitor A-804598 significantly prevented the effects of ATP on membrane and soluble CD163 expression (Figure 4c and 4d). In addition, pretreatment of ATP-stimulated M-MoDM with the metalloproteinase inhibitor TAPI-0 significantly increased the percentage of CD163-positive cells, i.e. 28.8 ± 43.4% (ATP) and 57.3 ± 14.7% (TAPI-0 + ATP) and significantly reduced the release of sCD163, i.e. 151.4 ± 25 ng/ml (ATP) and 97.9 ± 30.7 ng/ml (TAPI-0 + ATP) (Figure 4e). In contrast, the Figure S1 indicates that increasing concentrations of UTP or HMGB1, which increased ADAM17 activity in other cell types, did not modulate CD163 expression in M-MoDM.
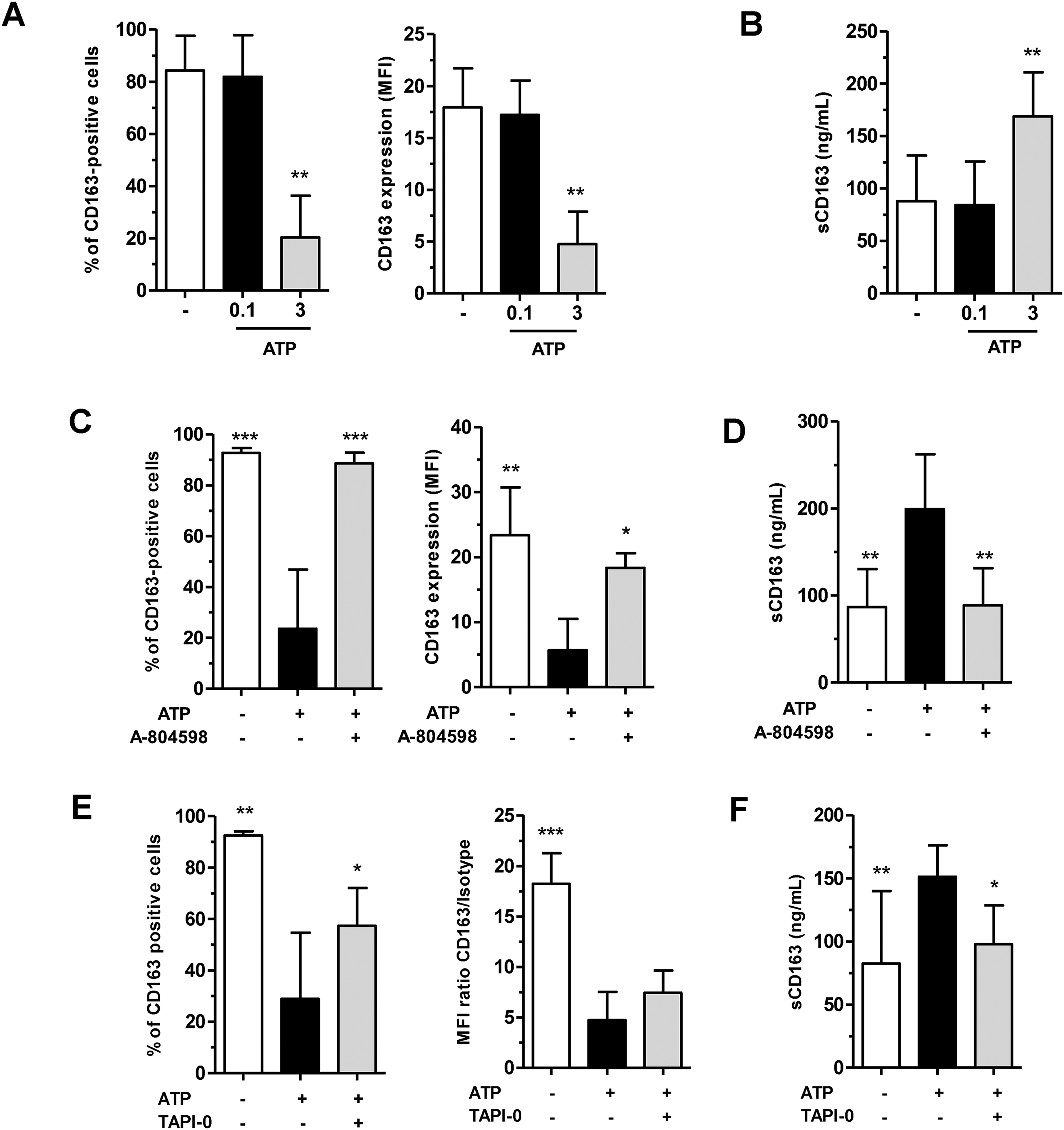
ATP modulated CD163 expression in M-MoDM. Macrophages were untreated (“-“) or pre-treated with A-804598 (c, d) or TAPI-0 (e, f) for 1 h before stimulation with ATP for 1 h. Membrane (a, c, e) and soluble (b, d, f) CD163 expressions were quantified by flow cytometry and ELISA, respectively. The results are expressed as the means ± SD of 3 (a), 5 (b, d) and 4 (c, e–f) independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus the corresponding (“-“) untreated cells in (A, B) and versus “ATP” in (c-f).
Polarization of M-MoDM modulated basal CD163 expression
We next determined if the polarization of M-MoDM (M0 status) into M1 and M2 macrophages could affect the membrane and soluble CD163 expression. The basal membrane CD163 expression (MFI, 34.8 ± 10.8) was significantly reduced in M1 (MFI, 25.2 ± 3.6) and increased in M2c MoDM (MFI, 49.5 ± 13.7), polarized in the presence of IFN-γ and IL-10 + dexamethasone, respectively, when compared to those measured in their M0 counterparts (Figure 5a, left panel). In addition, the basal sCD163 concentration (72.7 ± 35.9 ng/ml) was significantly increased in the M1 (140.9 ± 90.2 ng/ml), but not in the M2c (62.6 ± 28.4 ng/ml) MoDM culture medium when compared to the levels measured in the M0 culture medium (Figure 5a, right panel). LPS significantly diminished the intensity of the membrane CD163 expression in M1 and M2a, but not in M2c (Figure 5a). In contrast, LPS only significantly increased the secretion of sCD163 from M2c (62.6 ± 28.4 versus 237 ± 130.5 ng/ml) (Figure 5b).
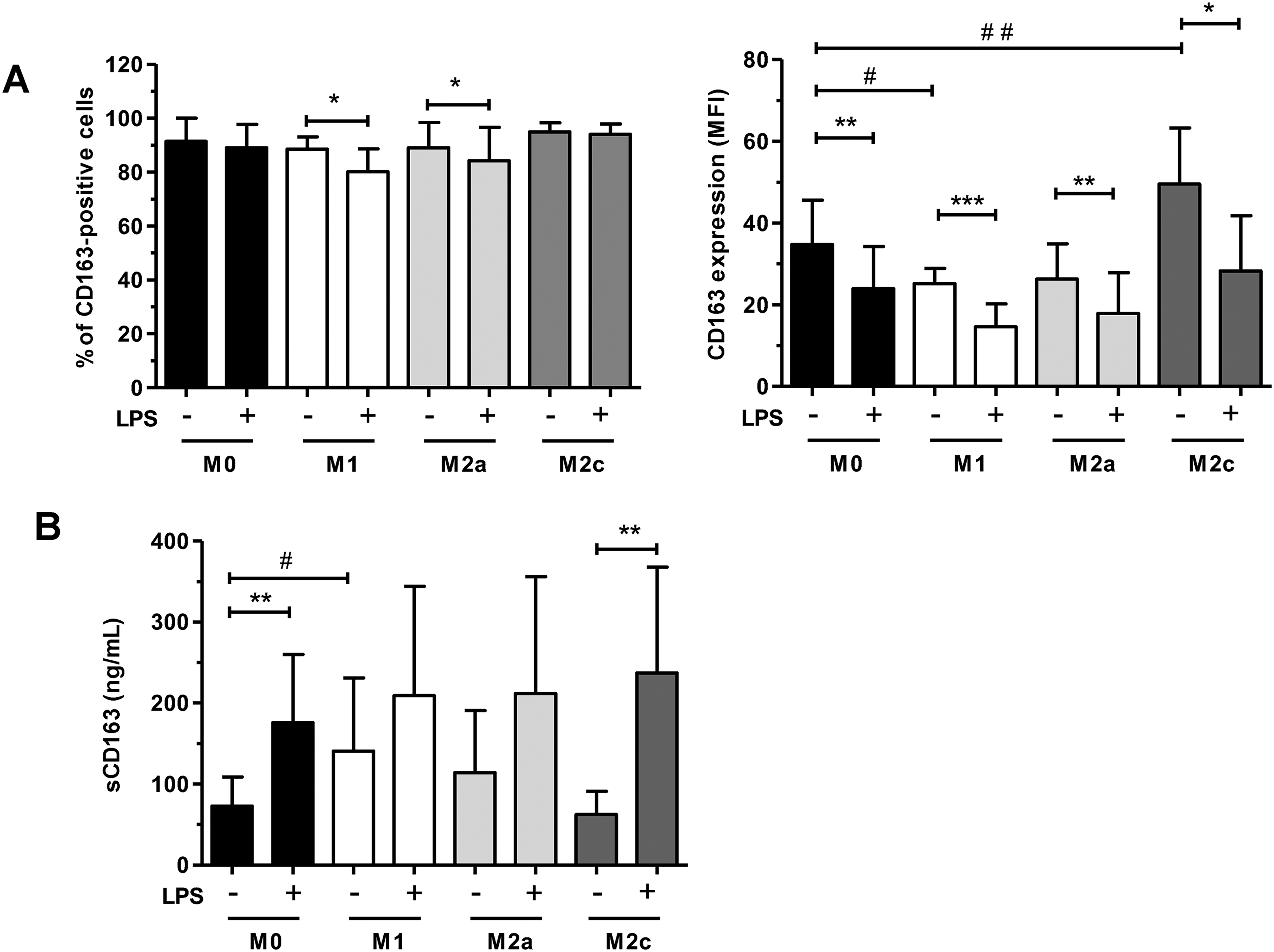
Membrane and sCD163 expressions in polarized M-MoDM. M-MoDM were left untreated (M0) or polarized into M1, M1 and M2c macrophages as described in materials and methods. Membrane (a) and soluble (b) CD163 expressions were quantified by flow cytometry and ELISA, respectively. The results are expressed as the means ± SD of 6 independent experiments. *, # P < 0.05, **, # #P < 0.01, ***P < 0.001.
Pulmonary CD163 expression in patients with IPF
In the next part of this study, we investigated the pulmonary CD163 expression in patients with IPF. Using immunofluorescence, we first studied the membrane CD163 expression on macrophages present on surgical lung biopsies taken from 6 patients with IPF. We found that, in each biopsy, several cells positive for the macrophage markers CD68 and CD206, thus defined as macrophages, also expressed membrane CD163 (Figure S2). CD163-positive macrophages are notably detected in the alveolar spaces. Thus, we next determined if sCD163 could be detected in BALFs. Figure 6a shows that sCD163 could be quantified in BALFs isolated from controls and patients with IPF or other ILD. In addition, basal sCD163 concentrations could be detected in culture media of alveolar macrophages isolated from each BALF (Figure 6b). Constitutive secretion of sCD163 from IPF alveolar macrophages (67.6 ± 44.6 ng/µg RNA) was significantly higher than the sCD163 release from control (19.2 ± 7.6 ng/µg RNA) and other ILD (31.5 ± 16.6 ng/µg RNA) alveolar macrophages. Stimulation with LPS significantly increased the sCD163 levels in culture media of the three groups of alveolar macrophages (Figure 6b). Alveolar macrophages also constitutively secreted TNF-α. However, the concentrations of sCD163 and TNF-α secreted by IPF or other ILD alveolar macrophages were not correlated (Figure 6c). Finally, we analyzed the CD163 expression in M-MoDM prepared with peripheral blood monocytes collected from 11 patients with IPF. LPS significantly enhanced the secretion of sCD163 from the IPF M-MoDM, i.e. 29 ± 20.5 ng/ml (M0) and 123.6 ± 72.8 ng/ml (LPS) (Figure 6d).
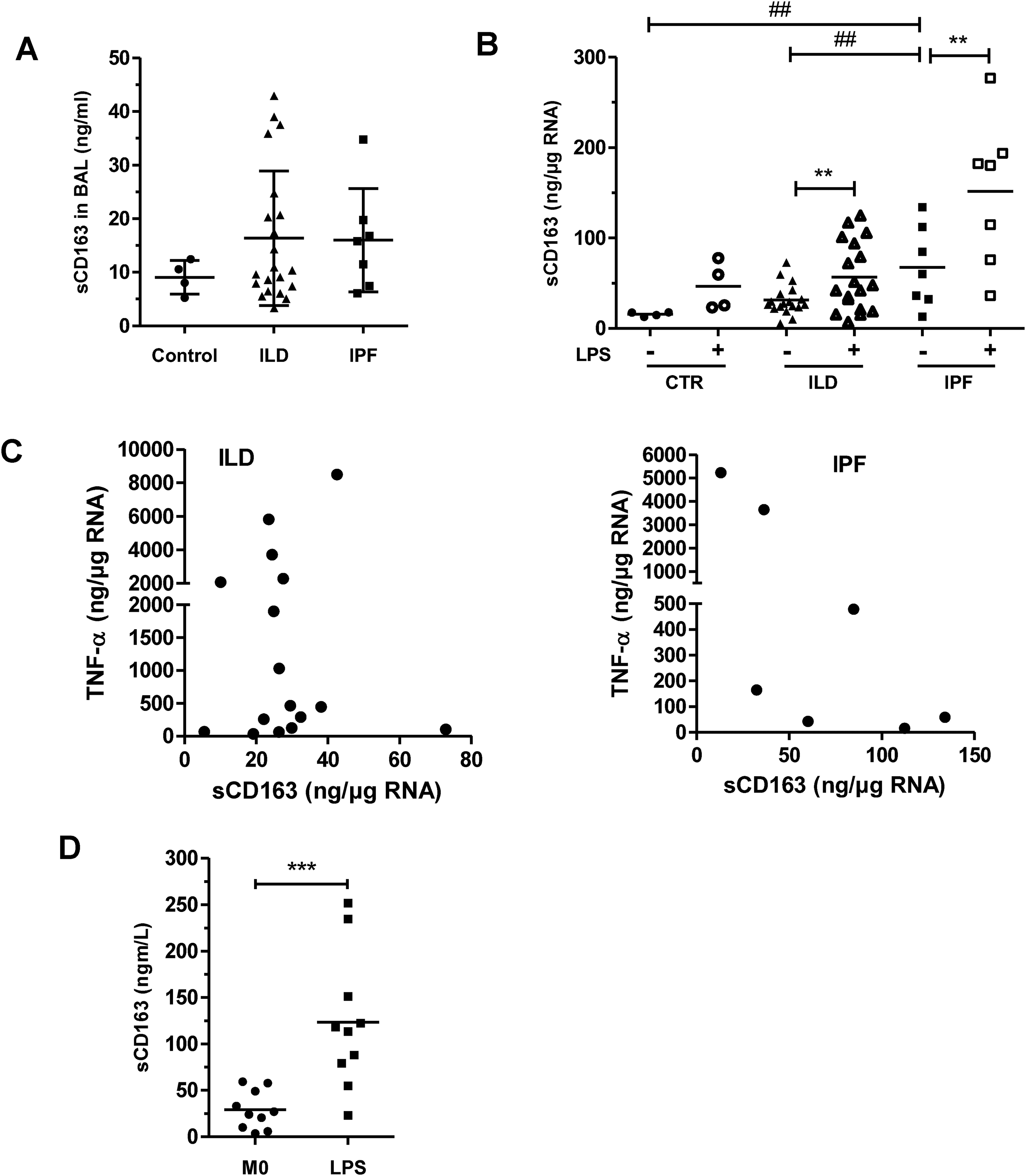
LPS increased the secretion of sCD163 from IPF alveolar macrophages and IPF M-MoDM. sCD163 concentrations were quantified by ELISA in BALFs (a) and culture supernatants from alveolar macrophage (b, c) or M-MoDM (d) isolated from controls or patients with IPF or other ILD. (c) TNF-α concentrations were also quantified by ELISA in culture supernatants from alveolar macrophage. Cells were left untreated or were stimulated with 20 ng/ml LPS for 24 h. In (b, c), the concentrations of cytokines secreted by macrophages were normalized to the respective cellular RNA concentration. *P < 0.05, **, # #P < 0.01, ***P < 0.001.
Serum sCD163 concentrations did not correlate with clinical parameters from patients with IPF
In the last part of our study, we measured the sCD163 concentrations in serum samples collected, at diagnosis, from 155 patients with IPF. We next studied the potential correlations between the values of the sCD163 concentrations and various functional and clinical parameters. No significant correlation could be demonstrated between the values of sCD163 and those of lung functional parameters (FVC and DLco values, % predicted) (data not shown). Similarly, serum sCD163 concentrations were not correlated with the numbers of blood monocytes or CRP concentrations in these patients (data not shown). Among the 155 patients, 96 were still alive at the end of the present study, whereas 59 were deceased or transplanted (Table 1). The FVC and DLco values (% predicted) were significantly decreased in the deceased or transplanted patients. In addition, we found that male gender, FVC and DLco (% predicted) constitute independent variable in the multivariate regression Cox model. In contrast, sCD163 concentrations were quite similar in the serum samples collected from the two groups of patients (Table 1).
Discussion
We first demonstrated in the present study that CD163 expression was strongly induced by M-CSF during macrophage differentiation. Indeed, M-MoDM constitutively displayed higher membrane levels of CD163 and secreted more amounts of sCD163 than circulating blood monocytes or GM-MoDM. Furthermore, we showed that LPS markedly increased the secretion of sCD163 from M-MoDM. We did not decipher the molecular mechanism controlling the shedding of membrane CD163 and the coordinated release of sCD163. However, the activation of some metalloproteases likely contributes to these processes since the pan-metalloprotease inhibitor TAPI-0 totally blocked the effects of LPS. TAPI-0 also prevented the secretion of sCD163 from blood monocytes 24 and GM-MoDM 25 stimulated with LPS or PMA. ADAM17 has been demonstrated to mediate the shedding of CD163 in CD163-overexpressing HEK293 cells. 26 Moreover, the tissue inhibitor of metalloproteinase-3 (TIMP-3), which more specifically represses ADAM17 activity, strongly prevented the release of sCD163 from phorbol myristate acetate (PMA)-stimulated GM-MoDM. 25 Other inhibitors such as TIMP-1, TIMP-2 and TIMP-4, that do not inhibit ADAM17 activity, did not block PMA effects on GM-MoDM. 25 Consequently, ADAM17 likely controlled the shedding of membrane CD163 in GM-MoDM stimulated with PMA. In contrast, this specific desintegrin and metalloproteinase may not control the release sCD163 from M-MoDM since the total inhibition of ADAM17 by genetic silencing did not prevent the modulation of CD163 expression by LPS even if TNFα secretion was inhibited, confirming the efficacy of gene silencing. Thus, such differences in sCD163 release mediated by ADAM17 may depend on cell types and/or stimulating agents.
ATP is a potent activator of metalloproteases controlling the shedding of different ectodomains of integral membrane receptors. 27 Patients with IPF, and mice developing lung fibrosis in response to bleomycin, have elevated concentrations of ATP in BALFs. 28 Interestingly, we found that, like LPS, ATP induced the coordinated decrease of membrane CD163 expression and increase of sCD163 release from M-MoDM. Its effects were also prevented by TAPI-0 suggesting that metalloproteases controlled the shedding of membrane CD163 in ATP-stimulated M-MoDM. ATP promotes inflammation and cell-mediated immunity mainly through activation of the P2X7 receptor. P2X7 receptor-deficient mice display a reduced expression of the main profibrotic factors, suggesting a role for P2X7 in lung fibrosis. 28 We showed that the P2X7 receptor, which is expressed on the plasmatic membrane of human M-MoDM, 29 likely mediated the ATP-dependent release of sCD163 from M-MoDM since the selective P2X7 receptor inhibitor A-804598 blocked the effects of the purinergic nucleotide.
Beside LPS and ATP stimulation, the polarization of M-MoDM into M1 or M2 subtypes also modulated CD163 expression. Indeed, the polarization of MoDM into M2c macrophages with IL-10 + dexamethasone, slightly but significantly, increased the intensity of membrane CD163 expression, likely by upregulating the CD163 gene transcription, as previously described. 1 However, M2c polarization did not enhance the basal release of sCD163. In addition, we show that the M1 polarization by IFN-γ decreased the levels of membrane CD163 in macrophages. Buechler et al. 30 previously reported that a 18-h treatment with IFN-γ similarly repressed CD163 expression at the macrophage membrane. IFN-γ suppresses IL-10 expression and consequently can indirectly decrease the CD163 mRNA levels controlled by this interleukin.30,31 Beside the regulation of CD163 gene transcription, IFN-γ could also stimulate the shedding of membrane CD163 as it significantly increased the basal secretion of sCD163 from M-MoDM (Figure 5).
We report in this study that CD68 and CD206-positive macrophages present in the pulmonary tissues of Caucasian patients with IPF could also express CD163. These cells are mainly located in the alveoli and may secrete sCD163 in alveolar fluids. Indeed, we detected sCD163 in BALFs taken from patients with IPF and we measured constitutive sCD163 levels in supernatant of alveolar macrophages isolated from these BALFs. It is noteworthy that basal sCD163 secretion of sCD163 from IPF alveolar macrophages was significantly higher than the sCD163 release from control or other ILD alveolar macrophages. The fact that the values of sCD163 and TNF-α concentrations were not correlated did not support a role for ADAM17 in mediating the basal secretion of sCD163 from IPF alveolar macrophages. Our results show that the constitutive sCD163 secretion from control alveolar macrophages was very low. This is concordant with the results reported by Gibbons et al. 15 who, using immunochemistry, showed that CD163-positive macrophages are systematically present in BALFs isolated from patients with IPF and nearly absent in control BALFs. LPS, which promotes lung fibrosis in different murine models,32,33 also significantly increased the secretion of sCD163 from alveolar macrophages. Considering both the role of M-CSF in the regulation of CD163 expression and the increased concentrations of M-CSF in BALFs from patients with IPF, 13 it can be hypothesized that CD163-positive macrophages detected in IPF lungs may derive from blood monocytes which differentiated into M-MoDM in the pulmonary tissues. Such CD163-positive macrophages present in pulmonary tissues of IPF patients may contribute to the pathophysiology of the disease. Indeed, the genetic invalidation of M-CSF expression and the pharmacological inhibition of M-CSF signaling decrease the presence of alveolar MoDM and ameliorate experimental lung fibrosis induced by bleomycin or asbestos fibers in mice.13,16 In addition, the selective genetic deletion of alveolar MoDM after their recruitment to the lungs also significantly reduce lung fibrosis in murine models. 14
The possibility that CD163 could be used as a relevant biological marker for IPF progression remains nevertheless to be demonstrated. First, it is still unclear if the numbers of CD163-positive macrophages quantified on lung tissues can predict the severity of the disease. To our knowledge, this potential correlation has not been studied in a cohort of Caucasian patients and the results of the two Asian studies that investigated this link are discordant.18,19 In addition, our results demonstrate that the serum concentrations of sCD163 were not correlated to the values of functional lung parameters measured in patients with IPF. As previously reported in two Asian studies performed on smaller cohorts (n = 90 and n = 60),20,21 our study indicates that the serum CD163 concentration was not an independent variable predicting the survival of Caucasian IPF patients in the multivariate Cox regression models. This negative result may be explained by the fact that serum sCD163 is probably produced by different types of monocyte/macrophages such as circulating monocytes and diverse peripheral tissue macrophages. Consequently, the part of serum sCD163 originating from lung macrophage may be too weak to increase significantly the total serum sCD163 concentration in patients with IPF.
In summary, our results demonstrate that in vitro the coordinated membrane CD163 expression and sCD163 release were significantly higher in human M-MoDM than in monocytes and GM-MoDM. The secretion of sCD163 from M-MoDM was stimulated by LPS and ATP, and was blocked by the metalloproteinase inhibitor TAPI-0. The polarization of M-MoDM into M1 macrophages in the presence of interferon-γ (IFN-γ) simultaneously reduced the basal expression of membrane CD163 and increased the release of sCD163. In addition, we report that, ex-vivo, alveolar macrophages isolated from patients with IPF secreted stronger amounts of sCD163 than alveolar macrophages isolated from controls and patients with other ILD. However, our results demonstrated that serum sCD163 concentrations were not correlated with the progression of IPF in the cohort of 155 Caucasian patients with IPF, which thus suggests that serum sCD163 could not constitute a predictive biomarker for the severity of this major ILD.
Footnotes
Abbreviations
Acknowledgements
We thank the Rennes Biosit platform for flow cytometry analysis. We thank Roselyne Viel from H2P2, Histopathological platform (Univ Rennes) for immunofluorescence analysis. We acknowledge Cécile Daoudal (ILD nurse, Centre Hospitalier Universitaire, Rennes) and the Centre de Ressources Biologiques (CRB) Santé of Rennes BB-0033-00056 for managing patient samples.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Pr Stéphane Jouneau has received fees, funding or reimbursement for national and international conferences, boards, expert or opinion groups, research projects over the past 5 years from Actelion, AIRB, Astra Zeneca, BMS, Boehringer Ingelheim Pharma, Chiesi, Gilead, GSK, LVL, Mundipharma, Novartis, Pfizer, Roche Pharma, Savara-Serendex. The other authors declare no competing interests.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article. This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), the Université de Rennes (Univ Rennes), Boehringer Ingelheim Pharma and Roche.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.