
Abstract
CD36 expressed in multiple cell types regulates inflammation, vascular function, and innate immunity. Specifically, CD36 in microvascular endothelial cells (ECs) signals to elicit inflammation and causes EC death. This study investigated roles for EC-CD36 on acute stroke pathology in normal and obese conditions. Obesity induced by a high-fat diet (HD) selectively increased CD36 expression in ECs, not in monocytes/macrophages, in the post-ischemic brain. Mice deficient CD36 in ECs (ECCD36−/−) showed reduced injury size and vascular permeability in normal conditions. While control mice fed a HD developed obesity and aggravated stroke injury, ECCD36−/− mice were resistant to develop an obesity phenotype. Subjecting ECCD36−/− mice to stroke resulted in reduced injury size and BBB disruption. Moreover, the mice had reduced MCP-1 and CCR2 gene expression, resulting in reduced monocyte trafficking with improved survival and acute motor function. Reduced MCP-1 and CCR2 expression was still evident in ECCD36−/− mice subjected to severe stroke, suggesting that monocyte trafficking is an infarct-independent metabolic effect associated with specific EC-CD36 deletion. Our findings demonstrate the importance of EC-CD36 in developing vascular comorbidities and suggest that targeting EC-CD36 is a potential preventative strategy to normalize vascular risk factors, leading to improved acute stroke outcomes.
Introduction
Stroke activates multiple pathological cascades that converge into inflammation and vascular dysfunction. The disruption of blood-brain barrier (BBB) integrity increases vascular permeability and promotes the infiltration of immune cells through paracellular and transcellular routes in the injured brain. The elicited neuroinflammation develops vasogenic edema, hemorrhagic transformation, and infarction.1,2 Previously, we reported that vascular risk factors such as diabetes and obesity aggravate vascular dysfunction and disproportionally increase brain edema in stroke. Attenuating the vascular permeability improved acute stroke outcomes in these comorbid conditions,3–6 suggesting that targeting vascular permeability may have potential to as a therapeutic strategy.
CD36 is a glycoprotein that is expressed in numerous cell types including monocytes/macrophages and microvascular endothelial cells (ECs). Through interaction with a host of ligands, it is a receptor involved in regulating innate immunity, inflammation, lipid uptake, and angiogenesis.7–10 As a scavenger receptor, CD36 expressed in monocytes/macrophages is engaged in the innate host response, 7 the most evolutionarily conserved function. In stroke, CD36 contributes to acute inflammation and infarct development. Mice with global deletion of CD36 (CD36 KO) have attenuated inflammation and are protected from brain injury and scar formation.6,9,11 Moreover, transplant of CD36-deficient bone marrow into control mice decreased stroke-induced MCP-1/CCR2 expression.9,11 Despite the inflammatory nature of macrophage CD36, CD36-mediated uptake of cellular debris and phagocytosis, contributing to tissue repair, 12 showed a context dependent role for macrophage CD36 in inflammation and resolution in the post-ischemic brain.
CD36 has high affinity for thrombospondin-1 (TSP-1); ligand-receptor interactions in the vessel wall induced signaling cascades, leading to MCP-1 and free radical production, BBB dysfunction and endothelial cell death.8,13,14 EC-CD36 has also been shown to contribute to stroke pathology. For example, stroke-elicited inflammation required the synergistic action of macrophage CD36 and the host response through ECs. 6 EC-CD36 promoted neutrophil neurotoxicity through CSF3 and was involved in IL-1β-induced endothelial activation, contributing to post-ischemic brain injury.10,15 In the hypertensive condition, a known risk factor for cerebrovascular disease, CD36 expression in vessels was associated with BBB impairment, 13 indicative of a potential role for EC-CD36 in the development of vascular pathology.
The purpose of this study is to define a cell type specific role for EC-CD36 in stroke pathology in normal chow and diet-induced obese conditions. Here we report that EC-CD36 contributes to the development of obesity, a vascular comorbidity. The contribution of EC-CD36 in stroke pathology include greater infiltration of monocytes, independent of infarct size and metabolic conditions. Resistance to development of obesity and improved stroke outcomes in EC-CD36 deficient mice suggest that selective targeting of EC-CD36 may serve as a potential strategy to normalize vascular risk factors that ultimately leads to improved stroke outcomes.
Material and methods
Animals
Procedures for the use of animals were approved by the Institutional Animal Care and Use Committee (IACUC) of Weill Cornell Medical College and carried out in accordance with guidelines from the National Institutes of Health, and Animal Research: Reporting of In Vivo Experiments (ARRIVE). To address the role of EC-CD36 in stroke, Floxed CD36 mice (CD36Flox) in the C57Bl/6j background were created by targeting exon 2 (exon 3 was included due to proximity), which contains the translation start site and first transmembrane domain and then crossed to EC-specific Tie2e Cre mice (ECCD36−/−). 16 CD36Flox and ECCD36−/− mice were bred and housed at Burke Neurological Institute Animal Facility. The facility monitors and maintains the temperature, humidity and 12 h light/dark cycle. A maximum of five mice were housed in a single cage with an individual ventilating system and irradiated bedding (The Anderson, Maumee, OH). Sterilized food and water were freely accessible in each cage.
Diet-induced obese mice
8-week-old mice were fed a high-fat diet (HD; 60% fat (S-3282), Bioserv, Flemington, NJ) for either 8 (males) or 12 weeks (females). Since female mice were resistant to diet-induced obesity, HD intervention was extended for female mice to obtain a similar degree of obesity between the sexes. Body weights (BW) were measured weekly. One week before the end of diet intervention, glucose tolerance tests (GTT) were performed in overnight fasted mice. Mice received an intraperitoneal injection of D-glucose (2 mg/g BW), and blood glucose levels were measured at 0 (baseline), 15, 30, 45, 120, and 180 min using a glucometer (Ascensia Contour; Bayer, Whippany, NJ).
Transient middle cerebral artery occlusion (MCAO)
Mice were subjected to transient MCAO as previously described. 9 Briefly, mice were anesthetized with isoflurane and connected to a Laser-Doppler Flowmeter (Periflux System 5010; Perimed) after a fiber-optic probe was glued to the parietal bone (2 mm posterior and 5 mm lateral to the bregma) for continuous monitoring of cerebral blood flow in the ischemic territory. For MCAO, a 6–0 Teflon-coated black monofilament surgical suture (coating length, 5–6 mm; diameter, 0.25 mm; Doccol, Redland, CA) was inserted into the exposed external carotid artery, advanced into the internal carotid artery, and wedged into the Circle of Willis to obstruct the origin of the MCA. The filament was advanced until a sufficient drop in cerebral blood flow (CBF) was observed. Reperfusion was confirmed at the time of filament withdrawal. All the studies in normal chow diet (ND) and HD-fed mice used 30 or 40 min transient MCAO. The CBF was measured before the stroke, during the occlusion period, and 10 min after reperfusion. Animals exhibiting greater than 80% reduction of pre-ischemic baseline CBF (20% of pre-stroke baseline) during MCAO and greater than 80% of pre-stroke baseline at 10 min of reperfusion were included in the study. Accordingly, 9.2% of animals were excluded from this study due to insufficient CBF reduction and/or reperfusion. Mice were randomized to a specific diet and surgery. Animals’ identity and treatment were blinded to the persons who assessed stroke outcomes.
Brain acute injury size measurement
Three days after MCAO, brains were isolated, cryosectioned at 20 μm thickness, and collected serially at 600 μm intervals. Infarct size and brain swelling measurements were performed using a total of 13 brain sections. Collected brain sections were examined with phase-contrast microscopy to visualize the infarcted area. Infarct territory was traced in each section to calculate the infarct area using ImageJ software. Infarct volume was determined by multiplying the infarct area (mm2) in each section by the distance (0.6 mm) to the next section and adding the results from all the sections. Infarct volume was corrected for edema by subtracting the hemispheric difference. To calculate percent hemispheric swelling, the difference between ipsilateral and contralateral hemispheric volume was divided by contralateral hemisphere volume and multiplied by 100.
Neurological assessment
Neurological deficits in motor and reflex skills were assessed based on motor and sensory tests 1d and 3d post-stroke. Neurological scores were graded using a scale of 0 to 14 (0, no deficits; 14, maximum deficits). 17 The motor function was tested by: 1) raising the mouse by its tail and evaluating forelimb flexion, hindlimb flexion, and head movement (>10° concerning the vertical axis within 30 sec; scored 0–3); 2) placing a mouse on the floor to determine the inability to walk straight, circling, and falling to the paretic side (scored 0–3); 3) abnormal movements to assess immobility and staring, tremor, and seizures, myoclonus or myodystony (scored 0–3). Sensory deficits were determined by a placing test and a proprioceptive test (scored 0–2). All post-stroke neurological assessments were carried out by investigators blinded to the animal’s genotype.
Open field test
To perform a motor function assessment, animals were placed in a 0.4 × 0.4 m square field surrounded by a 0.4 m high wall and allowed to freely explore the platform for 10 min. Total travel distance covered by the mice was tracked by analyzing live video with ANY-maze software (Stoelting Co., Wood Dale, IL). Center travel distance was used as a test for anxiety-related behavior.
mRNA measurement
Relative mRNA levels were quantified with real-time reverse transcription-polymerase chain reaction (RT-PCR) using fluorescent TaqMan technology. Total RNA samples from the contralateral and ipsilateral hemispheres, excluding the olfactory bulb and cerebellum, were extracted using Tri reagent (Miltenyi Biotec) and 500 ng of total RNA was reverse transcribed using M-MLV Reverse Transcriptase (Life Technologies, Foster City, CA), according to the manufacturer’s protocol. PCR primers and probes specific to the genes in this study were obtained as TaqMan pre-developed assay reagents for gene expression (Life Technologies, Foster City, CA). Primers used as follow; CCR2 (Mm00438270_m1), MCP-1 (Mm00441242_m1), CX3CL1 (Mm00436454_m1), CX3CR1 (Mm00438354_m1), CCL5 (Mm01302427_m1), CCR5 (Mm01963251_s1), and β-actin (Mm00607939_s1). β-actin was used as an internal control for the normalization of samples. The PCR reaction was performed in 20 μl total volume using FastStart Universal Probe Master Mix (ROX; Roche, Indianapolis, IN) by incubating at 95°C for 10 min followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C. The results were analyzed using 7500 Fast Real-Time PCR System software (Life Technologies, Grand Island, NY). To analyze the mRNA level, a delta-delta Ct (cycle threshold) method based on untreated samples and housekeeping gene (β-actin as internal control) was used. The overall formula to calculate the relative fold gene expression level was 2−ddCt (ddCt = dCt (treated sample) – dCt (untreated sample), and dCt (delta cycle threshold) =Ct (gene of interest) – Ct (β-actin)).
Flow cytometry analysis
For analysis of CD36 expression from brain tissues, brain homogenates were put through CD31− microbeads (Miltenyi Biotec, San Diego, CA) to enrich endothelial subset according to previously reported (Ref. your aflibercept paper here). CD31+ and CD31− population were used to analyzed for respective EC and monocyte/macrophage populations. Cells were incubated with antibodies (anti-mouse REA) against CD11b (PE-Vio770), CD45 (Vio-blue), Ly6C (APC), CD36 (FITC) and a mixture of phycoerythrin-conjugated antibodies (PE; Lin) against T cells (CD90.2), B cells (CD45R/B220), natural killer cells (NK-1.1 and CD49b), and granulocytes (Ly6G) in PBS for 30 min in the dark. After washing with PBS, cells were immediately analyzed with a MACS Quant VYB flow cytometer (Miltenyi Biotec). For circulating immune cell analysis, total CD45+ cells were determined by fluorescence triggering of CD45+ event read to reduce the background due to numerous CD45 negative cells in the blood. Antibody specificity was determined by analyzing cell-only, single, and double antibody controls for validation. All antibodies for flow cytometry were purchased from Miltenyi Biotec and used at a 1:50 ratio.
Vascular permeability assay
For IgG immunohistochemistry, frozen brain sections (20 μm) were fixed with 4% paraformaldehyde (PFA) for 15 min, incubated with 3% H2O2 for 30 min to block endogenous hydroperoxides (HRP), incubated in a blocking solution containing 1% bovine serum albumin (BSA) and 10% normal goat serum for 1 h, and labeled with anti-mouse IgG antibody (1:1000, Vector Lab., Burlingame, CA) overnight at 4°C. After washing with PBS, the sections were incubated with avidin/biotinylated peroxidase (ABC reagent, Vector Lab., Burlingame, CA) for 1 h at room temperature and colorized using the diaminobenzidine (DAB) (Sigma-Aldrich) system. IgG+ area and optical density (OD)were quantified using ImageJ software. To account for the non-specific staining, IgG intensity in the IgG+ area in the ipsilateral side was expressed as a ratio of the contralateral hemisphere.
CD36 protein measurement
Total protein concentrations were measured by the Bradford method. Protein was loaded on a NuPAGE 4-12% Bis-Tris Gel (Life Technology) and transferred onto a polyvinylidene fluoride membrane (Bio-Rad, Hercules, CA). The membrane was incubated in blocking buffer (Li-cor, Lincoln, NE) for 1 h followed by overnight incubation at 4°C with anti-CD36 (1:1000, Cell signaling, MA) or anti-β-Actin (1:10,000, Santa Cruz Biotechnology, Santa Cruz, CA) antibody in blocking buffer. The membrane was washed with tris-buffered saline containing 0.05% Tween-20 followed by incubation with appropriate secondary antibodies conjugated with IRDye® 680RD (Li-cor) in blocking buffer for 1 h and visualization using the Odyssey Imaging System (Li-cor). To normalize inter-blot variability, identical samples were loaded on each blot as an internal control and the density of the internal standard sample was used to standardize other samples in multiple blots.
Statistics
All data are expressed as the mean ± S.D. The sample size for in vivo studies was determined based on predicting detectable differences to reach the power of 0.80 at a significance level of <0.05. Student’s t-test was used for comparing differences between 2 groups and multigroup analyses were performed using two-way ANOVA. Bonferroni post hoc comparisons of the ANOVA were used to compare either genotype or stroke effects using Prism 9.4. To determine the survival curve, a log-rank test was performed. The normality of data was analyzed using the Kolmogorov-Smirnov and Shapiro-Wilk tests for all data. In cases where data did not show normal distribution, Mann–Whitney U test was utilized. The level of significance was set at p < 0.05.
Results
Obesity augments stroke-induced CD36 expression in EC, but not monocyte-derived macrophages (MMØ)
In our previous study, we demonstrated that stroke increased CD36 expression in the injured brain, and this increase was further augmented in hyperlipidemic mice, exacerbating acute stroke outcomes. 18 Given that obese mice also displayed adverse stroke outcomes, 4 we determined whether CD36 is involved in the exacerbation of stroke in obesity. Stroke-induced CD36 gene expression in the ipsilateral hemisphere was comparable between lean and obese mice (Figure 1(a)), but CD36 protein levels were significantly higher in obese mice (1.55 ± 0.62 vs 2.69 ± 0.92; lean vs obese, p < 0.01) (Figure 1(b)), showing obesity augments CD36 protein in the post-stroke brain.
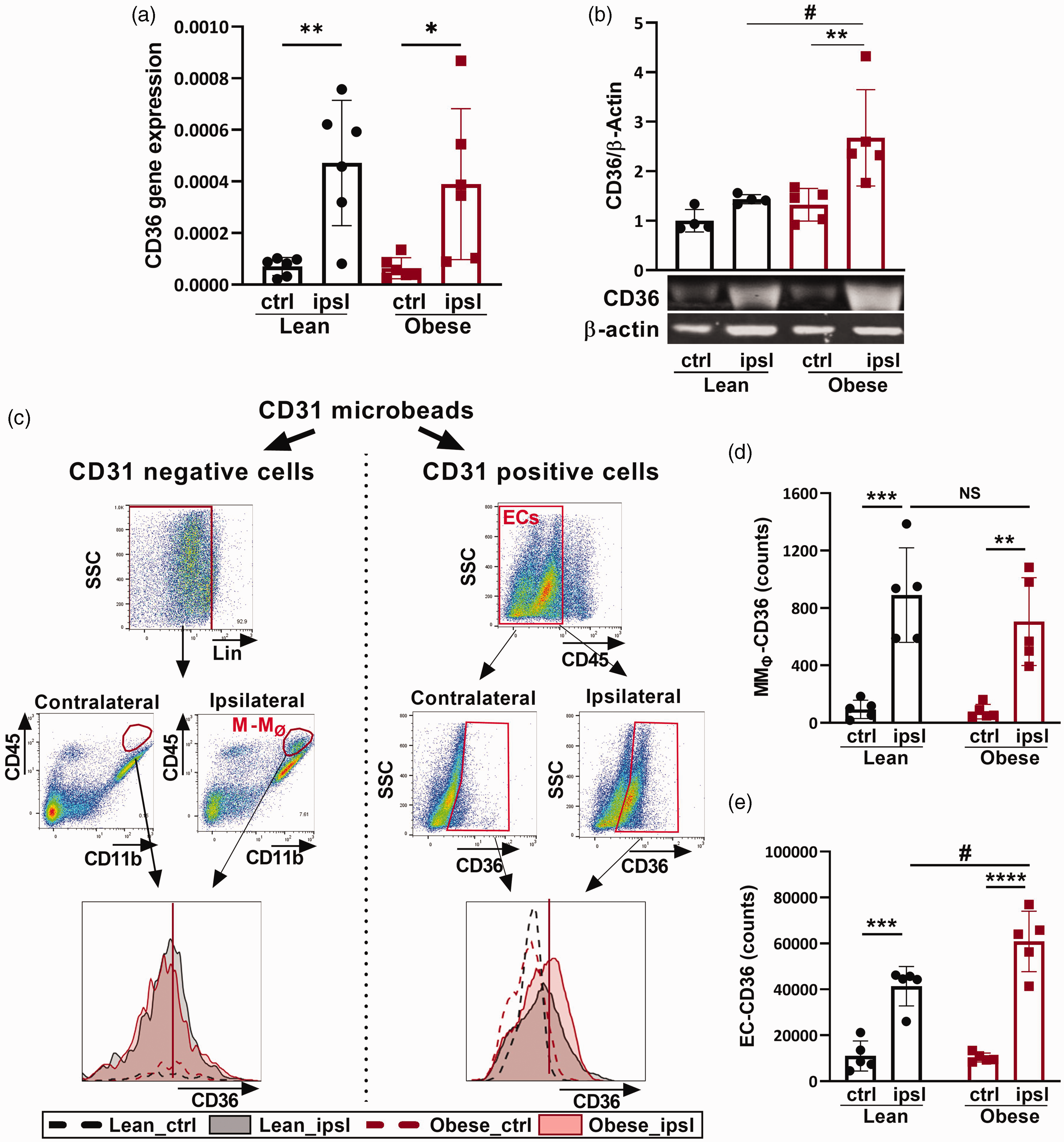
Stroke-induced CD36 expression in lean and obese mice. Mice were subjected to transient MCAO for 30 min. (a, b) Assessment of CD36 gene and protein expression in the brain at 3d postischemia. (c) Gating strategy to define M-MØ (monocytes/macrophages) and ECs (endothelial cells) populations at 3d post-ischemia. CD31− and CD31+ populations were obtained from isolated brain cells by passing through CD31 magnetic bead columns. MMØ are defined as [CD31−/CD11b+/CD45+/Lin−]. Lin is the combined linage markers for NK (NK1.1), neutrophils (Ly6G), B (CD45R), and T cells (CD90.2). ECs are defined as [CD31+/CD45−] cells. (d, e) Quantification of CD36+ cells expressed in MMØ and ECs. ctrl, contralateral; ipsl, ipsilateral; Data are mean ± SD; Two-way ANOVA, *, **, ***, ****p < 0.05, 0.01, 0.001, 0.0001 vs ctrl, #p < 0.05 vs lean.
CD36 function varies depending on interactions with ligands in specific cell types. To address which cell type accounted for the obesity enhanced CD36 expression in the post-stroke brain, we determined CD36 protein levels in MMØ and ECs. Using CD31 microbeads, isolated brain cells were separated into CD31− cells for MMØ and CD31+ cells for ECs (Figure 1(c)). From these populations, the number of CD36+ cells was determined by flow cytometry by gating on the CD31−/CD11b+/CD45High/Lin− (lineage marker for T, B, NK cells and granulocytes) population for MMØ and the CD31+/CD45− population for EC, as EC do not express CD45 (Figure 1(c)). Stroke-induced MMØ-CD36 expression in the ipsilateral hemisphere of obese mice was similar to lean mice (1.9 × 104 ± 0.8 × 104 vs 1.8 × 104 ± 0.8 × 104; lean vs obese, ns) (Figure 1(d)). Further analysis revealed that CD36 expression in microglia (CD45Low) and other infiltrated immune cells (combined NK cells, neutrophils, B and T cells (Lineage+; Lin+) were also not significantly different between lean and obese mice (Supplementary Fig. 1). CD36 expression on EC was also significantly increased in the ipsilateral hemisphere both of lean and obese mice compared to the contralateral, however, the number of cells induced in obese mice was significantly greater (4.1 ± 0.9 vs 6.1 ± 1.3 cells (104), p < 0.01) (Figure 1(e)). These results indicated that obesity-enhanced CD36 expression in stroke is mainly accounted for by increased EC expression, not MMØ, and led us to investigate the mechanistic role of EC-CD36.
Endothelial CD36 deficiency reduces stroke-induced brain injury
To determine the role of EC-CD36 in stroke, we generated mice with conditional deletion of CD36 in ECs (ECCD36−/−). 16 Compared to control (CD36Flox) mice, ECCD36−/− mice showed a selective reduction of stroke-induced CD36 expression in ECs but not in MMØ, validating the model for a specific CD36 deletion in ECs (Figure 2(a)). The number of CD36+ cells in the contralateral side was low but the number was similar between genotypes (Figure 2(a)). However, we observed a significant reduction in CD36 expression level, indicated by the reduction of CD36 mean fluorescence intensity (MFI) in ECCD36−/− mice (6.0 ± 0.8 vs 4.6 ± 0.6, CD36Flox vs ECCD36−/−, n = 5/group, p < 0.05) supporting that conditional deletion of CD36 in ECs reduces baseline CD36 expression in ECs. To address the underlying event by which EC-CD36 contributes to acute stroke outcomes, infarct size and brain swelling were assessed at 3d post-MCAO in transgenic mice. Compared to control mice, ECCD36−/− mice displayed attenuated stroke-induced infarct size (35.9 ± 15.8 mm3 vs 17.4 ± 3.2 mm3, p < 0.01) and hemispheric brain swelling (22.3 ± 7.1% vs 10.3 ± 6.5%, p < 0.0001) (Figure 2(b)). The better acute stroke outcomes were accompanied by reduced vascular permeability, indicated by IgG immunostaining. Careful examination revealed that ECCD36−/− mice had smaller IgG+ area (9.9 ± 3.8 mm2 vs 5.2 ± 3.1 mm2, p < 0.01) and reduced intensity of IgG extravasation (15.1 ± 4.5 AU vs 5.2 ± 4.8 AU, p < 0.01) (Figure 2(c)). These findings collectively indicated that EC-CD36 contributed to stroke-induced acute brain injury and BBB permeability.
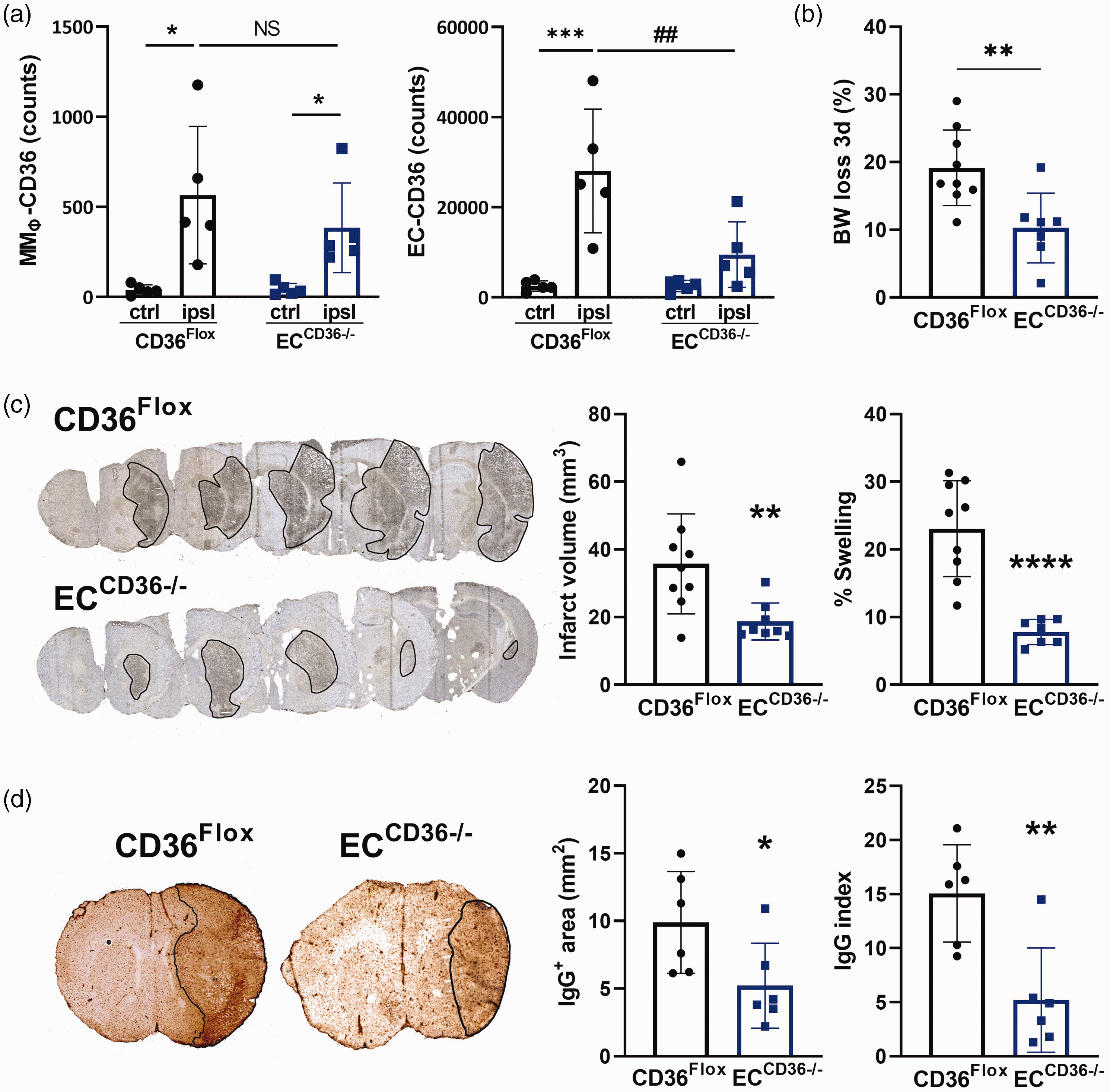
Effect of CD36 deletion in ECs on stroke outcome. (a) Stroke-induced CD36 expression in ECs and MMØ in CD36Flox and ECCD36−/− mice. n = 5/group; ctrl, contralateral; ipsl, ipsilateral. Data are mean ± SD; Two-way ANOVA, *, ***p < 0.05, 0.001, vs ctrl, ##p < 0.01 vs CD36Flox. (b) BW loss at 3 days post-ischemia. Presented as percentage of pre-ischemic value (% Pre). (c) Infarct volume and hemispheric brain swelling at 3d post-MCAO. (d) Assessment of vascular permeability by IgG immunostaining. Quantification of IgG+ area and average IgG intensity of IgG+ area. Data are presented as mean ± SD; Student’s t-test, *, **, ****p < 0.05, 0.01, 0.0001 vs CD36Flox.
ECCD36−/− mice resist the development of obesity and have reduced stroke severity
To investigate the role of EC-CD36 in stroke in the context of obesity, experimental obesity was induced by feeding CD36Flox and ECCD36−/− mice a high fat diet (HD) for 8 (males) or 12 (females) weeks, such that a similar obese phenotype was obtained for both sexes. In addition to the anti-angiogenic role of EC-CD36, CD36 expressed in ECs regulates tissue fatty acid uptake and controls whole body metabolism.16,19 Consistently, in CD36Flox mice, HD intervention resulted in rapid weight gain and the development of insulin resistance, as indicated by impaired glucose clearance (Figure 3(a)). HD-diet intervention did not produce an obese phenotype in ECCD36−/− mice (Figure 3(a)). Compared to CD36Flox mice, stroke outcome analyses in ECCD36−/− mice showed reduced infarct volume (47.2 ± 15.4 mm3 vs 27.5 ± 15.8 mm3, p < 0.001) and brain swelling (26.0 ± 13.9% vs 15.6 ± 13.7%, p < 0.05) and reduced vascular permeability (area, 16.1 ± 3.0 mm2 vs 4.9 ± 2.6 mm2 p < 0.0001; index, 23.4 ± 6.0 AU vs 5.8 ± 3.1 AU, p < 0.0001) (Figures 3(b) and (c)). The results demonstrated that EC-CD36 contributed to obesity-enhanced brain injury and suggested that the effect was mediated by metabolic regulation.
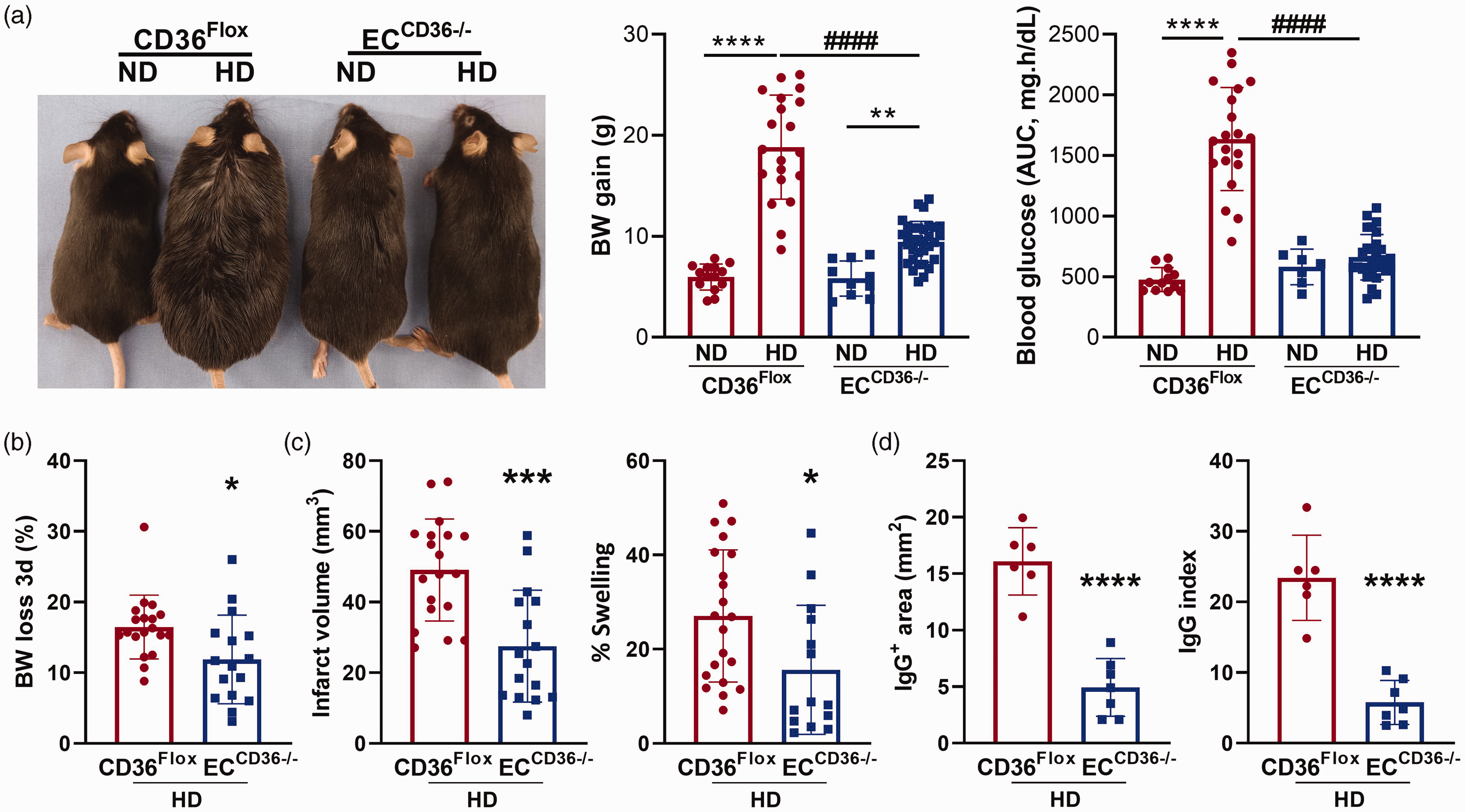
Effect of diet on acute stroke outcomes. (a) Representative images of CD36Flox and ECCD36−/− mice fed a normal (ND) or high fat diet (HD). Body weight (BW) gain and area under the curve (AUC) derived from glucose tolerance testing (GTT) over 180 min, were evaluated in week 7. Data are presented as mean ± SD; Two-way ANOVA, **, ****p < 0.01, 0.0001 vs. normal-diet (ND), ####p < 0.0001 vs. CD36Flox. (b–d) Assessment of stroke outcomes in HD fed CD36Flox and ECCD36−/− mice for body weight loss (% pre-ischemic) (b), infarct volume and swelling (c), and vascular permeability, IgG+ area and IgG index (d). Data are mean ± SD; Student’s t-test, *, **, ****p < 0.05, 0.01, 0.0001 vs CD36Flox.
Stroke-induced monocyte infiltration is reduced in ECCD36−/− mice
Entry of circulating monocytes into inflamed tissue requires the interaction between MCP-1, a chemokine released from damaged tissue, and its cognate receptor, CCR2, expressed on inflammatory monocytes. In a previous study, we found that global deletion of CD36 (CD36 KO mice) reduced MCP-1 and CCR2 expression in the post-ischemic brain.9,18 To address the role played by EC-CD36 on monocyte trafficking, we compared MCP-1 and CCR2 expression between CD36Flox and ECCD36−/− mice fed the ND. Stroke increased the expression of MCP-1 after 3 days in both CD36Flox and ECCD36−/− mice, but the expression in ECCD36−/− mice was significantly less (Figure 4(a)). While stroke induced increased expression of CCR2 in CD36Flox mice, the upregulation was absent in ECCD36−/− mice (Figure 4(b)). The attenuation of MCP-1 and CCR2 expression in ECCD36−/− mice was selective because the expression of other chemokines and receptor genes including CX3CL1, CX3CR1, CCL5, and CCR5 did not show genotype-dependent differences (Supplementary Fig. 2). Monocyte trafficking, indicated by an increase in the CD45High subset in brain after acute stroke,20–22 was significantly reduced in ECCD36−/− mice (2430 ± 575 vs 983 ± 224 cells, p < 0.05) (Figure 4(c)).
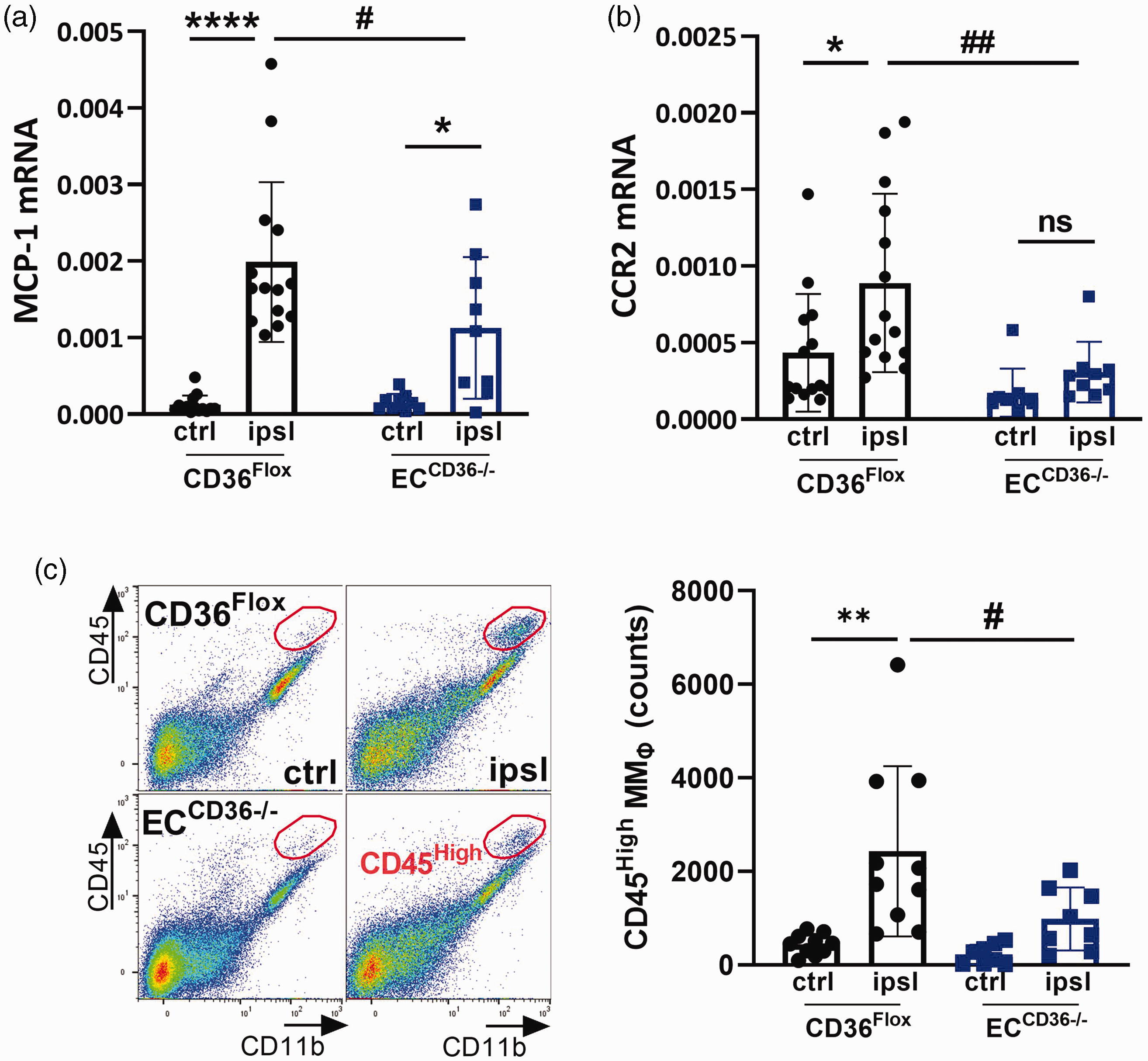
Effect of EC-CD36 deletion on monocyte trafficking. (a, b) Assessment of MCP-1 and CCR2 gene expression in CD36Flox and ECCD36−/− mice at 3d post-MCAO and (c) Flow cytometry gating strategy to identify infiltrated monocytes [CD45High] in the ipsilateral hemisphere. Quantification of CD45High MMØ counts. ctrl, contralateral; ipsl, ipsilateral; Data are mean ± SD; Two-way ANOVA, *, **, ****p < 0.05, 0.01, 0.0001 vs. ctrl, #,##p < 0.01, 0.05, vs CD36Flox.
Because stroke severity is positively linked to the extent of monocyte infiltration, 23 there is the possibility that the smaller injury size in ECCD36−/− mice may result in reduced monocyte trafficking. We thus increased the duration of MCAO to 40 min in ECCD36−/− mice fed the ND to equalize infarct volume to be similar to CD36Flox mice with 30 min MCAO. This approach produced similar infarct size between the genotypes (Figure 5(a)). Despite similar degree of brain injury, vascular permeability and MCP-1/CCR2 gene expression were attenuated in the ipsilateral hemisphere of ECCD36−/− mice (Figure 5(b) and (c)), supporting the infarct independent reduction of vascular permeability and MCP-1/CCR2 gene expression in the absence of CD36 in ECs. Similarly, ECCD36−/− mice fed the HD showed less MCP-1 and CCR2 gene expression (Figures 6(a) and (b)) and also significantly reduced monocyte trafficking (Figures 6(c)). Collectively, these results suggested the importance of EC-CD36 in monocyte trafficking, regardless of stroke severity and metabolic phenotype.
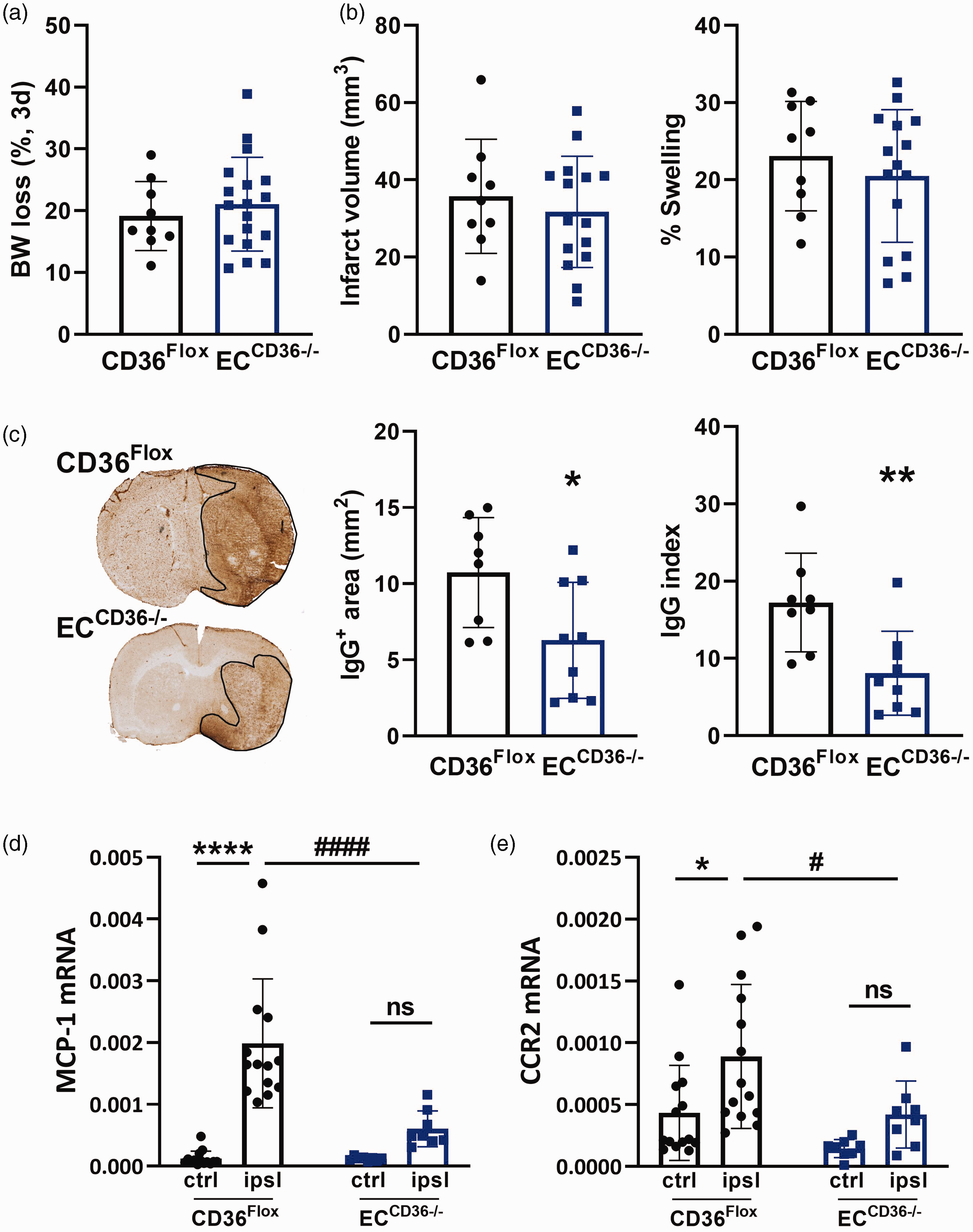
Effect of EC-CD36 deletion on stroke outcome and MCP-1 CCR2 expression in severe stroke in ND fed mice. CD36Flox and ECCD36−/− mice were fed ND and subjected to 30 min and 40 min MCAO respectively to produce similar infarct size between genotypes. (a, b) Assessment of body weight loss (%), infarct volume, and hemispheric brain swelling assessed at 3d post-MCAO. (c) Assessment of vascular permeability by IgG immunostaining. Quantification of IgG+ area and average IgG intensity of IgG+ area. Data are presented as mean ± SD; Student’s t-test, *, **p < 0.05, 0.01, vs CD36Flox. (d, e) Gene expression of MCP-1 (d) and CCR2 (e). ctrl, contralateral; ipsl, ipsilateral; Data are presented as mean ± SD; Two-way ANOVA, *, ****p < 0.05, 0.0001, vs ctrl, #,###p < 0.05, 0.001 vs CD36Flox.
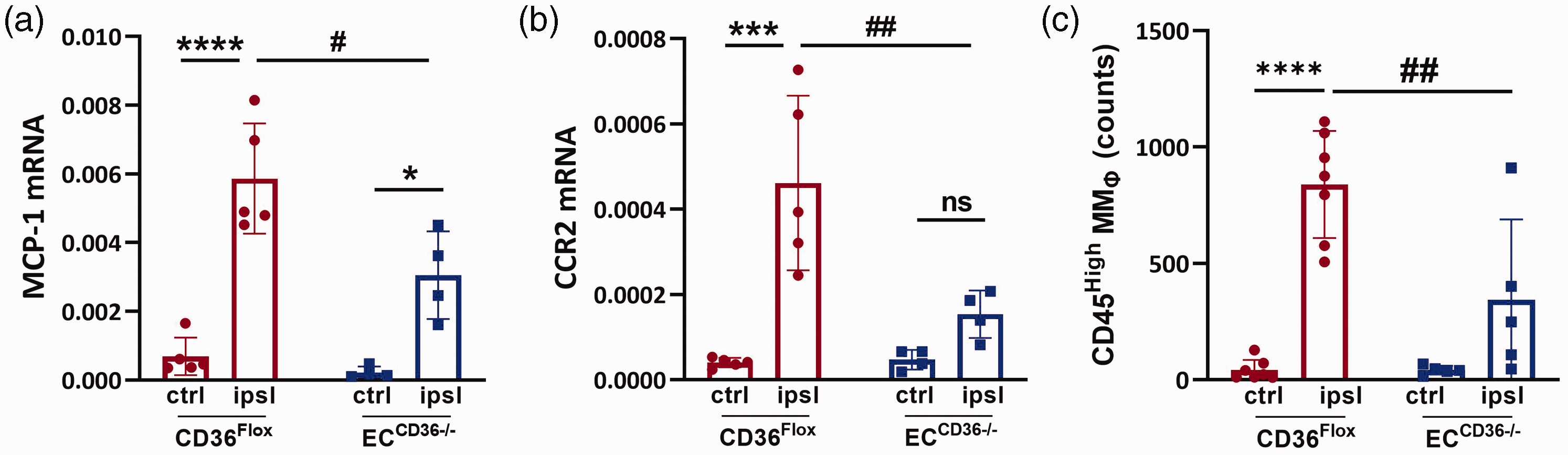
Effect of EC-CD36 deletion on monocyte trafficking in mice fed the HD. (a, b) MCP-1 and CCR2 gene expression at 3d MCAO in the brains of the HD-fed CD36Flox and ECCD36−/− mice. (c) Quantification of CD45High MMØ counts. ctrl, contralateral; ipsl, ipsilateral; Data are mean ± SD; Two-way ANOVA, *, ***, ****p < 0.05, 0.001, 0.0001 vs. ctrl, #, ##p < 0.01, 0.05 vs CD36Flox.
EC-CD36 deficiency reduced stroke-induced mortality and improved motor function
As monocyte trafficking predominantly occurs during the acute phase of stroke, post-stroke survival and neurological impairment at this period were assessed. In the ND-fed mice, there were no significant differences in post-stroke mortality between genotypes (19.0%, 4 out of 21 vs. 11.5%, 3 out of 26; CD36Flox vs. ECCD36−/−, ns) (Figure 7(a)). In mice fed the HD, CD36Flox mice displayed a high post-stroke mortality rate (46.1%, 18 died out of 39), but mortality was significantly reduced in ECCD36−/− mice (4.5%, 1 out of 22, p < 0.01) (Figure 7(c)). ECCD36−/− mice also had significantly reduced acute neurological severity score in both diet conditions (Figures 7(b) and (d)). Further assessment of motor deficit by an open-field test revealed that ECCD36−/− mice had significantly higher motor activity, covering greater total distance and center-distance within the 10 min test duration (Figure 7(e)). These results suggested contribution of EC-CD36 to acute neurological and motor impairment.
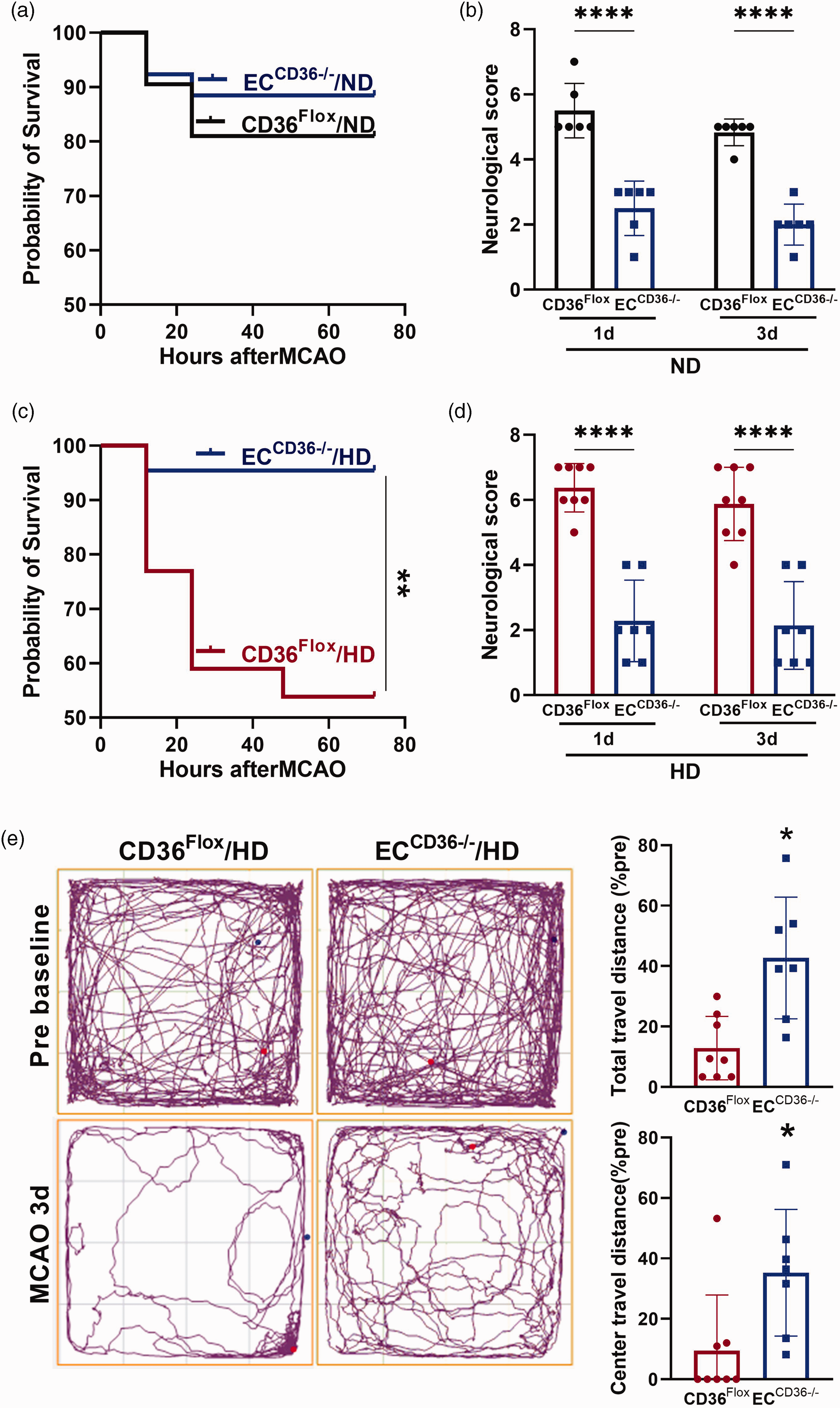
Effect of EC-CD36 deletion on acute neurological and motor deficits after MCAO. CD36Flox and ECCD36−/− mice fed ND (a, b) and HD (c–e) were subjected to 30 min MCAO. (a, b) Post-stroke survival rate and neurological impairment scores at 1 and 3 days after MCAO in ND-fed mice. (c) Post-stroke survival rate in HD-fed mice. (d, e) Neurological score, and open field test in HD-fed mice. Data are mean ± SD; Statistical tests: Log-rank test for survival rate, *p < 0.05 vs. CD36Flox; Two-way ANOVA test for neurological score; t-test for infarct size, swelling and total travel distance; Mann–Whitney U test for center travel distance; *, **, ****p < 0.05, 0.01, 0.0001 vs. CD36Flox.
Discussions
Cardio- and cerebrovascular risk factors including hyperlipidemia, diabetes, and obesity are prevalent among stroke patients. Our previous studies with the inclusion of these comorbidities in experimental brain ischemia showed exacerbated brain injury,4,6,9,18 demonstrating the importance of considering vascular risk factors for better understanding of stroke pathology and therapy development. In addition to a well-recognized role as a scavenger receptor, CD36 functions in transporting fatty acids into cells and is involved in developing vascular risk factors, including obesity/insulin resistance, and in regulating systemic metabolism.16,24,25 Thus, the current study addressed the role played by EC-CD36 in experimental brain ischemia, vascular permeability and monocyte trafficking in normal and obese conditions.
Previous studies reported that CD36 contributed to eliciting inflammatory responses, BBB dysfunction, and scar formation, and these events were accompanied with greater neurological deficits.9,11,12,26,27 These studies were mostly conducted in mice with global deletion of CD36; subsequent studies on the cell type specific role of CD36 in monocytes/macrophages demonstrated involvement in the pathogenesis of atherosclerosis and stroke-induced brain injury.6,28 Despite the detrimental effects caused by CD36 expression in macrophages, CD36-mediated uptake of apoptotic cells/cellular debris by MMØ in the injured tissue is critical to the resolving phase of inflammation and for tissue remodeling. Consistently, this reparative activity of CD36 expressed in phagocytic macrophages has been reported in stroke, 12 demonstrating a dichotomous role for CD36 in stroke pathology and recovery. The context-dependent role of CD36 has been attributed to its expression in specific cell types. While macrophage CD36 has been shown to provoke innate host responses,29,30 the inflammatory response in stroke is derived from CD36 expressed in ECs.10,15
Several reports have addressed the role of EC-CD36 in triggering EC inflammation and metabolic diseases by increasing fatty acid uptake, altering tissue lipid metabolism and leading to insulin insensitivity.16,19,31 Owing to its ability to transport long chain fatty acids, CD36 has an important physiological function. Studies utilizing cell type specific deletion of CD36 demonstrated that CD36 expressed in ECs, not cardiomyocytes, regulated and optimized fatty acid uptake into the heart. 19 In addition, EC-CD36 deletion had overall systemic metabolic effects. 16 This is consistent with our finding that mice with EC-CD36 deletion were refractory to the development of obesity and insulin resistance (Figure 3). The relevance of CD36 to vascular comorbidities was also reported for stroke-prone hypertensive animals. In these animals, CD36 expression in vessels was increased and this was associated with BBB impairment, 13 confirming the link between EC-CD36 and vascular pathology in a comorbid condition.
The severity of stroke, indicated by loss of BW, is closely related to the extent of monocyte infiltration. 32 CD45High/Ly6C+ MMØ significantly correlated with BW reduction/injury size in the stroked brain, while CD45Low/Ly6C+ MMØ did not. 32 Since a larger injury size results in higher immune cell infiltration, our observation that there was reduced MCP-1 and CCR2 expression, and less CD45High MMØ in the brains of ECCD36−/− mice may have been due to smaller infarct volume. We excluded this possibility by increasing the duration of MCAO in ECCD36−/− mice, in order to produce similar infarct size in both ECCD36−/− mice and controls. Despite similar infarct size, our results still showed significantly attenuated MCP-1 and CCR2 expression in ECCD36−/− mice (Figure 5), supporting an upstream metabolic effect of EC-CD36 deletion on the major chemokine-receptor expression affecting monocyte infiltration (Figure 4). Since CD36 activation in EC signals to produce the CC chemokine MCP-1, and causes EC apoptosis, deletion of EC-CD36 has overall benefit, resulting in less MMØ infiltration into the brain independent of infarct size.
To target CD36 in a comorbid condition, a previous study used salvianolic acid B (SAB) in naïve non-stroked mice, while feeding a HD. This pharmacological inhibitor of CD36 effectively attenuated visceral obesity and improved glucose clearance, leading to normalization of metabolic dysfunction and decreased adipose tissue inflammation. 32 In the current study, our observation that EC-CD36 deficient mice fed a HD were resistant to weight gain and the development of insulin resistance (Figure 3) indicated that systemic metabolic effects, through EC-CD36, is a predominate feature, and resembles CD36 global deletion. Despite the absence of the obese phenotype in ECCD36−/− mice, the current study defined the role of EC-CD36 in developing vascular comorbidity prior to stroke and its influence on vascular permeability and monocyte trafficking following stroke. A recent report showed a reduction in circulating Ly6CLow monocytes in EC-CD36 deletion, 33 however, we and others found no difference in circulating monocyte subsets in ECCD36−/− mice compared to control (Supplementary Fig. 3). 16 Thus, our study suggests that the EC-CD36 effect on stroke pathology occurs via MCP-1/CCR2-dependent monocyte trafficking, rather than the modulation of peripheral monocyte subsets.
In summary, the present study demonstrates that EC-CD36 contributes to the development of vascular comorbidities and stroke-induced brain injury in normal and obese conditions. An underlying mechanism is the regulation of MMØ infiltration through modulation of the MCP-1/CCR2 axis by EC-CD36. Resistance to HD-induced obesity and improved stroke outcomes in ECCD36−/− mice further suggests that targeting CD36 specifically in ECs may provide a potential strategy to normalize vascular risk factors, which could lead to acute neuroprotection and improved stroke outcomes.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X231154602 - Supplemental material for Endothelial cell CD36 mediates stroke-induced brain injury via BBB dysfunction and monocyte infiltration in normal and obese conditions
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X231154602 for Endothelial cell CD36 mediates stroke-induced brain injury via BBB dysfunction and monocyte infiltration in normal and obese conditions by Il-doo Kim, Hyunwoo Ju, Joseph Minkler, Roselyn Jiang, Abhilasha Singh, Roopa Sharma, Maria Febbraio, Sunghee Cho in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (NIH) grants R01NS103326, NS095359, and NS111568 (SC).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
IK participated in study design, data collection, statistical analyses, interpretation of data and wrote the manuscript, HJ participated in interpretation of the data and revised the manuscript. JM and RJ participated in behavior data collection. AS participated in molecular and biochemical assay of CD36. RS participated revised the manuscript. MF provided CD36Flox and ECCD36−/− mice, discussed project, and wrote the manuscript. SC participated in study design, statistical analyses, and interpretation of the data, and wrote the manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.