
Abstract
Acute-on-chronic liver failure (ACLF) carries a significant burden on critical care services and health care resources. However, the exact pathogenesis of ACLF remains to be elucidated, and novel treatments are desperately required. In our previous work, we utilized mice subjected to acute insult in the context of hepatic fibrosis to simulate the development of ACLF and documented the favorable hepatoprotection conferred by M2-like macrophages in vivo and in vitro. In the present study, we focused on the phenotypic switch of human and mouse macrophages and assessed the effects of this switch on apoptosis resistance in hepatocytes. For this purpose, human and mouse macrophages were isolated and polarized into M0, M(IFN-γ), M(IFN-γ→IL-4), M(IL-4) or M(IL-4→IFN-γ) subsets. Conditioned media (CM) from these subsets were applied to human and mouse hepatocytes followed by apoptosis induction. Cell apoptosis was evaluated by immunostaining for cleaved caspase-3. As a result, M(IFN-γ) or M(IL-4) macrophages switched their phenotype into M(IFN-γ→IL-4) or M(IL-4→IFN-γ) through reprogramming with IL-4 or IFN-γ, respectively. Importantly, hepatocytes pre-treated with M(IFN-γ→IL-4) CMs exhibited much weaker expression of cleaved caspase-3, compared to those pre-treated with M(IFN-γ) CM, and vice versa. Together, phenotypic switch of macrophages toward M(IL-4) phenotype confers hepatocytes enhanced resistance to apoptosis.
Keywords
Introduction
Acute-on-chronic liver failure (ACLF) occurs in the setting of chronic liver diseases, and exacerbates after precipitating events such as acute viral, drug, or alcoholic hepatic insults.1,2 In comparison, acute liver failure (ALF) occurs most often in patients who do not have pre-existing liver diseases.3,4 Subjectively thinking, ACLF patients would suffer from more severe hepatic damage than ALF patients. Instead, ACLF has a relatively lower mortality.5 This phenomenon coincides with the previous report that patients with prior hepatic decompensation have lower short-term mortality than those without prior decompensation.5,6 Herein, previous decompensation may be considered as a beneficial response instead of deleterious event. Moreover, there is a crucial “golden window” period preceding sepsis development and organ failure in ACLF, which provides the valuable opportunity for reversing progressive liver failure through therapeutic interventions.6,7 This motivates us to decipher the underlying mechanisms governing the favorable protection against acute insult in ACLF.
We and others have attempted to explore this issue. Considering that currently there is no canonical mouse model of ACLF, researchers utilize mice subjected to acute insult in the context of hepatic fibrosis to simulate the development of ACLF. In this regard, hepatic fibrosis induced by carbon tetrachloride (CCl4), bile duct ligation (BDL), and thioacetamide (TAA) has been demonstrated to exert the hepatoprotective effects against various acute insults including
As we all know, macrophage polarization is mainly modulated by local microenvironmental signals. Most importantly, macrophage activation is a highly dynamic process, and the phenotype of polarized macrophages can be reversed under physiological and pathological conditions.15–17 In this regard, modulation of macrophage activation to acquire desirable phenotype and function may provide promising therapies to promote tissue repair.18–21 To prove this supposition, our in vitro experiment will focus on the phenotypic switch of macrophages in response to different stimuli and assess the effect of this switch on hepatocyte apoptosis. To be specific, polarized human and mouse macrophages were reprogrammed, then conditioned medium experiments were conducted followed by apoptosis induction. The apoptosis resistance of human and mouse hepatocytes were compared before and after phenotypic switch of macrophages. Our findings help advance the understanding of the pathogenesis of ACLF and will shed light on a novel therapeutic intervention through manipulating macrophage polarization.
Materials and methods
Animals
Male BALB/c mice (6–8 wk old) were obtained from Laboratory Animal Center, Academy of Military Medical Sciences, Beijing, China. Mice were housed in a specific pathogen-free (SPF) environment at 22–24°C with alternatively 12 h light–dark cycles. Animals were fed standard laboratory chow with free access to water. All animal care and experimental procedures performed in this study were in accordance with the Guide for the Care and Use of Laboratory Animals and approved by Institutional Animal Care and Use Committee at Beijing YouAn Hospital affiliated to Capital Medical University.
Reverse transcription (RT) and SYBR Green real-time quantitative PCR (qPCR)8
Total RNA was extracted from isolated macrophages using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Reverse transcription of the purified RNA (2 μg) was performed using random primers and AMV retrotranscriptase system (TakaRa, Dalian, Liaoning, China) according to the manufacturer’s protocol. SYBR Green real-time PCR was carried out using the ABI StepOne Plus (Applied Biosystems, Foster City, CA, USA). All reactions were performed in triplicate. The primers used were designed with Primer 3.0 software and listed in Table 1. The relative expression of target genes was calculated and normalized to the expression of GAPDH, a housekeeping gene.
Primer sequences for real-time PCR.

Isolation and in vitro polarization of primary mouse macrophages
Primary mouse macrophages (see Figure 1) were isolated from the livers of mice by pronase (Roche Diagnostics GmbH, Mannheim, Germany) and collagenase (Sigma-Aldrich, St. Louis, MO, USA) digestion followed by differential centrifugation using our previously reported method.10 Isolated macrophages (non-polarized M0 macrophages) were stimulated with mouse recombinant IFN-γ (100 U/ml, PeproTech, Rocky Hill, USA) or IL-4 (10 ng/ml, PeproTech). Twenty-four h later, parts of M(IFN-γ) or M(IL-4) macrophages were exposed to IL-4 or IFN-γ stimulation, respectively, for another 24 h. In other words, mouse macrophages were divided into five groups: M0, M(IFN-γ), M(IFN-γ→IL-4), M(IL-4), and M(IL-4→IFN-γ). The phenotype of the subsets was identified through qRT-PCR analysis for gene signatures of representative markers.10,22–25 In addition, supernatants from all subsets were collected for conditioned medium experiment.
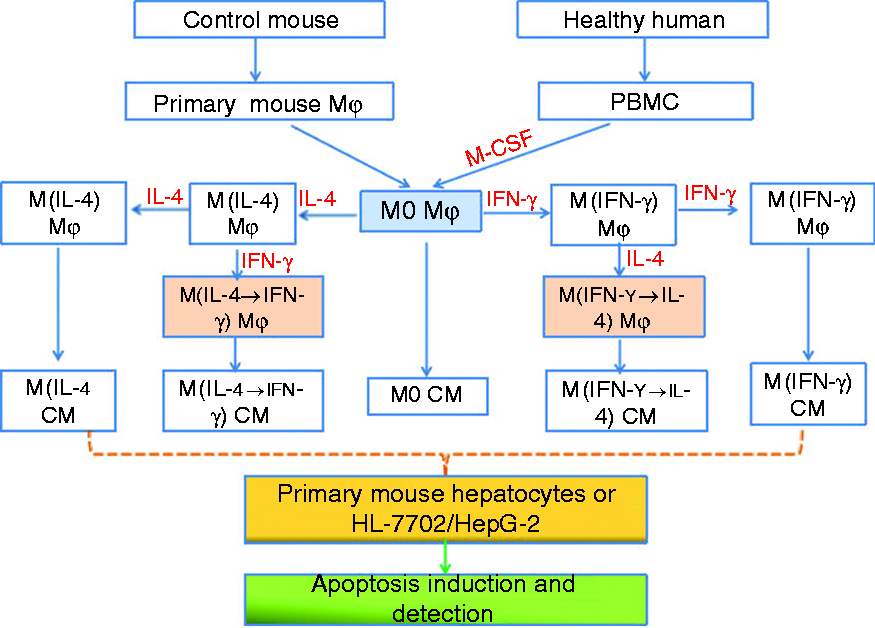
A schematic flowchart of the study design.
Isolation, culture, and polarization of human monocyte-derived macrophages
PBMCs were isolated from the whole blood of healthy donors and ACLF patients using a Ficoll (Hao Yang Biological Manufacture Co. Ltd., Tian Jin, China) density gradient, and cultured in DMEM supplemented with 10% FBS. Two h later, non-adherent cells were removed,26 and the resultant adherent cells (monocytes) were cultured in DMEM supplemented with human recombinant macrophage colony-stimulating factor (M-CSF, 50 ng/ml, PeproTech) for 6 d. Differentiated macrophages (non-polarized macrophages) were polarized with human recombinant IFN-γ (50 ng/ml, Peprotech) or IL-4 (50 ng/ml, Peprotech). Similarly to mice, parts of M(IFN-γ) or M(IL-4) macrophages derived from PBMCs of healthy donors were exposed to IL-4 or IFN-γ stimulation, respectively, for another 24 h. As mentioned above, human macrophages were also divided into five groups, and the phenotype of the subsets was identified through qRT-PCR analysis.10,14 Supernatants from all subsets were also collected for conditioned medium experiment.
Conditioned medium experiments and apoptosis detection
Conditioned media (CMs) from human and mouse macrophages, i.e., M0 CM, M(IFN-γ) CM, M(IFN-γ→IL-4) CM, M(IL-4) CM, and M(IL-4→IFN-γ) CM were collected and centrifuged to remove cell debris. Then, CMs from above-mentioned groups were incubated with primary mouse hepatocytes or human liver cell lines (HL-7702 and HepG2) for 6 h, and then cell apoptosis was induced by human and mouse TNF-α (50 μg/ml, Peprotech)/
Cell culture
Human liver cell lines (HL-7702 and HepG2), primary mouse hepatocytes and isolated mouse/human macrophages were cultured in DMEM (Gibco, Grand Island, NY, USA) supplemented with 10% heat-inactivated FBS (Gibco) and 1% penicillin/streptomycin (Gibco) in a 37°C incubator.
Statistical analysis
Results were expressed as mean ± SEM. Group comparisons were performed using Student’s t test or one-way ANOVA followed by Newman–Keuls multiple comparison test. Statistics and graphs were generated using Prism 5.0 software (GraphPad Software Inc., San Diego, CA, USA). P < 0.05 was considered statistically significant.
Results
The phenotypic switch of mouse macrophages in vitro
First, the phenotypic switch of murine liver macrophages was validated in vitro. Polarized M(IFN-γ) or M(IL-4) macrophages were reprogrammed with IL-4 or IFN-γ, respectively. Then, the representative markers of macrophage activation were analyzed by real-time PCR. The gene levels of M(IFN-γ) markers including TNF-α and iNOS were much higher in M(IFN-γ) macrophages, nevertheless, higher expression of M(IL-4) markers including YM-1, Arg-1, CD206, and TGF-β was noticed in M(IL-4) macrophages. That was to say, the polarization of macrophages was induced successfully. Importantly, remarkably elevated expression of M(IL-4) markers but reduced expression of M(IFN-γ) markers was found in M(IFN-γ) macrophages subjected to IL-4 stimulation [M(IFN-γ→IL-4) macrophages] compared to M(IFN-γ) macrophages, however, significantly down-regulated expression of M(IL-4) markers but up-regulated mRNA levels of M(IFN-γ) markers were detected in M(IL-4) macrophages subjected to IFN-γ stimulation [M(IL-4→IFN-γ) macrophages] compared to M(IL-4) macrophages (Figure 2). Thus, mouse macrophages were able to adapt and shape their phenotype in response to the microenvironmental change.
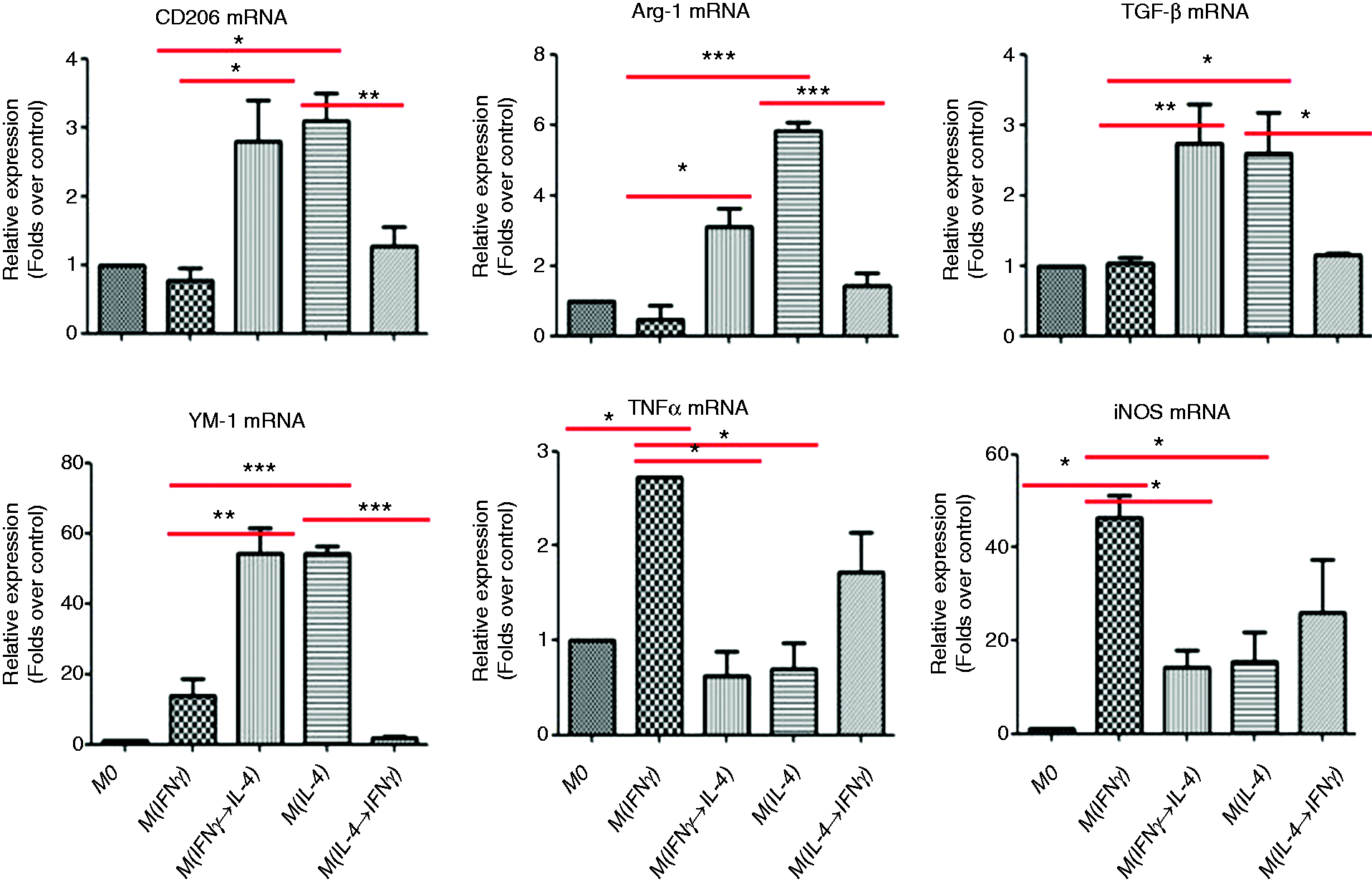
The phenotypic switch of primary mouse liver macrophages in vitro. Isolated primary mouse liver macrophages were polarized into M(IFN-γ) or M(IL-4) phenotype using IFN-γ or IL-4, respectively. Parts of M(IFN-γ) or M(IL-4) macrophages were reprogrammed with IL-4 or IFN-γ, respectively, to induce the phenotypic switch. Then, the representative markers of macrophage activation were analyzed by real-time PCR. Data were expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001
Skewing macrophages toward M(IL-4) phenotype confers primary mouse hepatocytes enhanced apoptosis resistance
In the previous work, we have demonstrated that M(IL-4) macrophages confer beneficial hepatoprotection through promoting M(IFN-γ) macrophage apoptosis but preventing hepatocyte apoptosis.10,14 To further validate the protective effects of M(IL-4) macrophages in mice, polarized M(IFN-γ) or M(IL-4) macrophages were reprogrammed into M(IFN-γ→IL-4) or M(IL-4→IFN-γ) phenotype, and the effects of phenotypic switch of macrophages on hepatocyte apoptosis were assessed by conditioned medium experiment followed by apoptosis induction. As a result of immunostaining, the expression of cleaved caspase-3, a canonical marker of cell apoptosis, was substantially reduced in primary mouse hepatocytes pre-treated with M(IFN-γ→IL-4) CM, compared to that in hepatocytes pre-treated with M(IFN-γ) CM. Nevertheless, apoptosis was remarkably enhanced in hepatocytes pre-treated with M(IL-4→IFN-γ) CM, compared to that in hepatocytes pre-treated with M(IL-4) CM (Figure 3). Therefore, phenotypic switch of macrophages toward an M(IL-4) phenotype confers mouse hepatocytes enhanced apoptosis resistance.
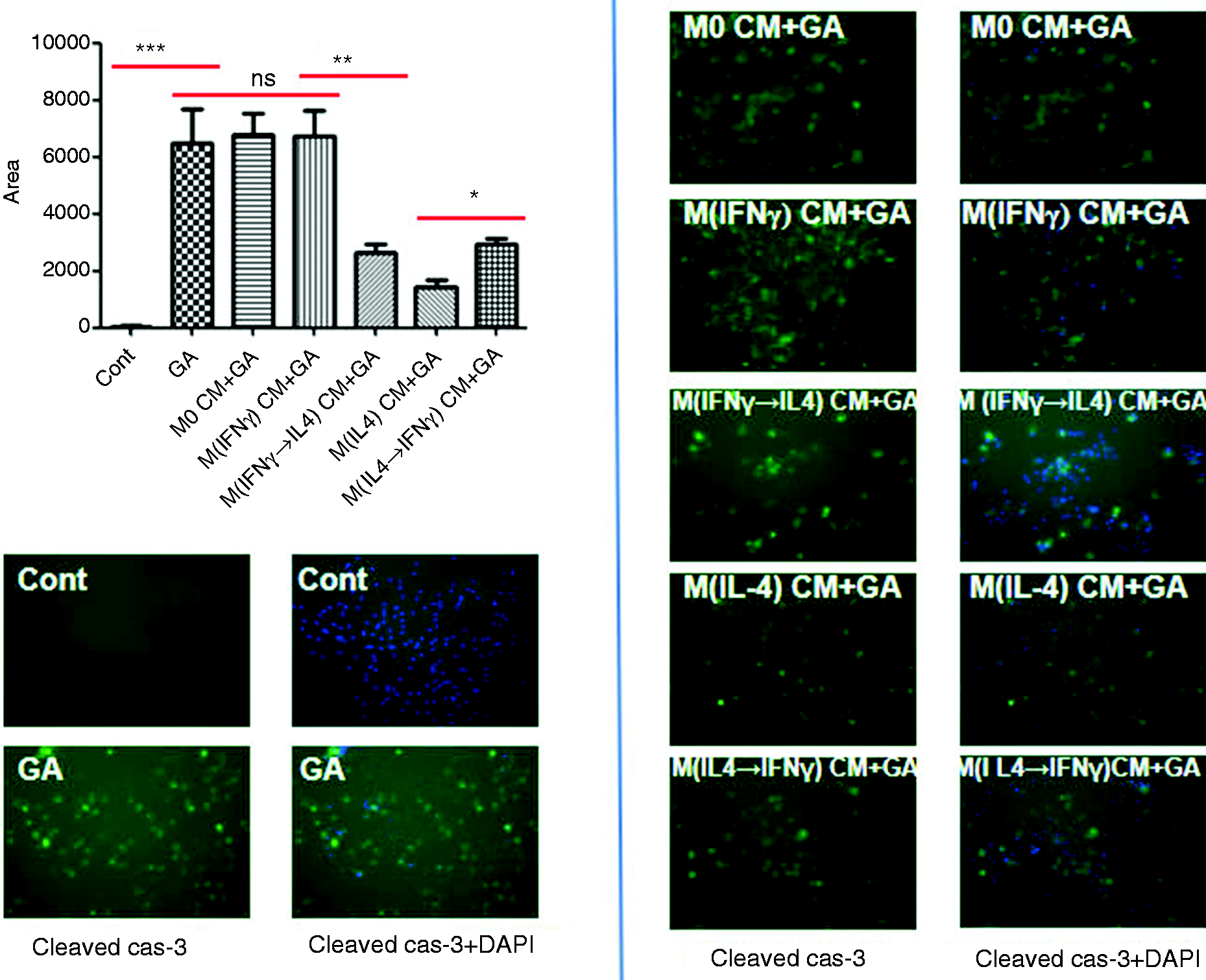
Skewing liver macrophages toward M(IL-4) phenotype confers primary mouse hepatocytes enhanced apoptosis resistance. Conditioned media from mouse M0, M(IFN-γ), M(IFN-γ→IL-4), M(IL-4), and M(IL-4→IFN-γ) macrophages were applied to primary mouse hepatocytes for 6 h. Cell apoptosis was induced by mouse TNF-α (50 µg/mL, Peprotech)/
The phenotypic switch of human macrophages in vitro
Isolated and differentiated human PBMC-derived macrophages were polarized into M(IFN-γ) or M(IL-4) macrophages, then stimulated with human IL-4 or IFN-γ, respectively, to induce phenotypic switch. The activation phenotype of resultant subsets was identified by real-time PCR. Similarly to mice, elevated expression of M(IL-4) markers (TGF-β and CTLA-1) but reduced expression of M(IFN-γ) markers (TNF-α and CD86) was found in M1-like macrophages subjected to IL-4 stimulation [M(IFN-γ→IL-4) macrophages] compared to M(IFN-γ) macrophages, however, significantly up-regulated mRNA levels of M(IFN-γ) markers (TNF-α and iNOS) but down-regulated expression of M(IL-4) markers (IL-10 and TGF-β) were detected in M(IL-4) macrophages subjected to IFN-γ stimulation [M(IL-4→IFN-γ) macrophages] compared to M(IL-4) macrophages (Figure 4). Thus, human PBMC-derived macrophages adapt their phenotype corresponding to microenvironmental cues.
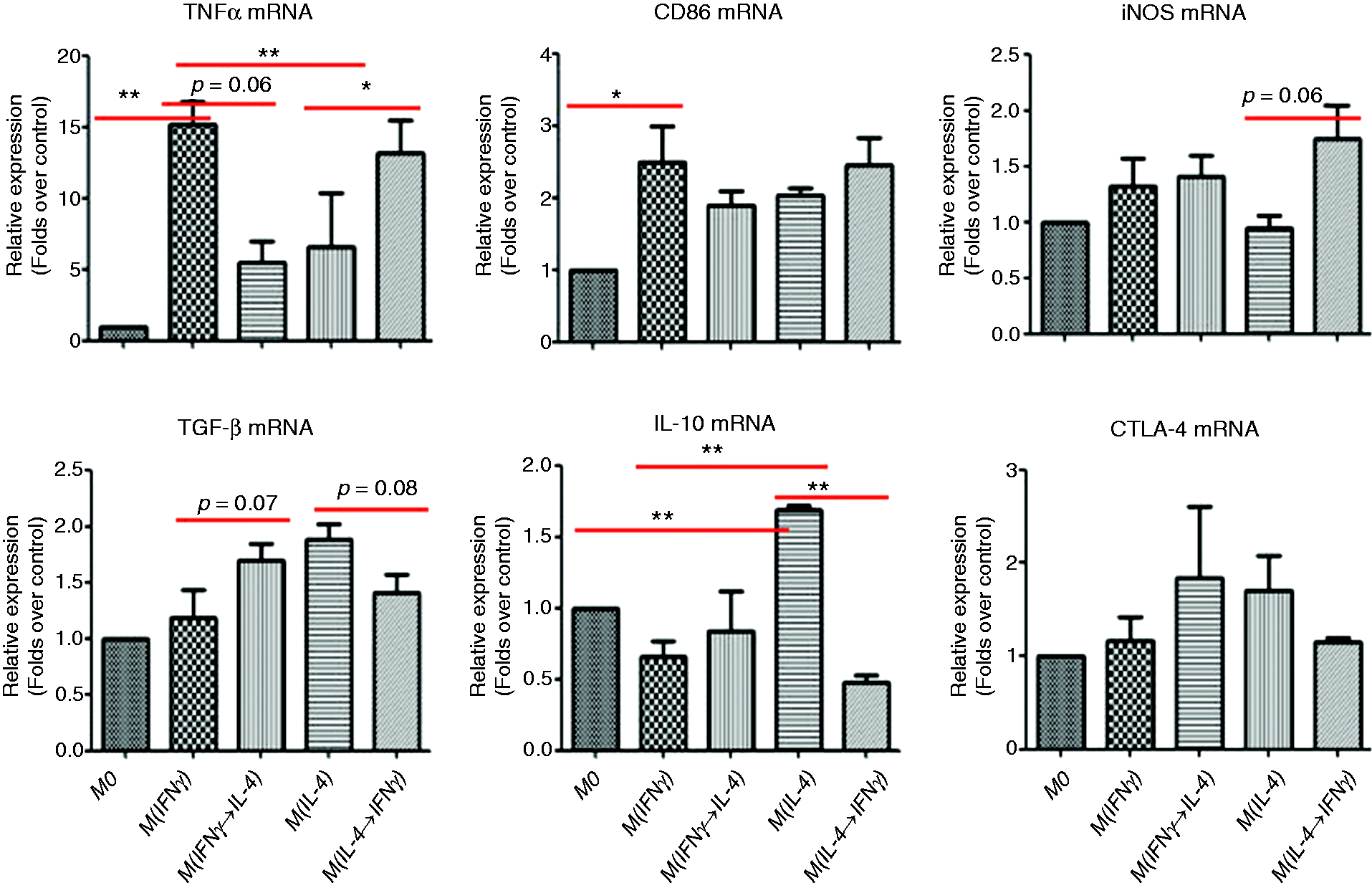
The phenotypic switch of human PBMC-derived macrophages in vitro. Human monocyte-derived macrophages were isolated from PBMCs of healthy donors, then polarized into M(IFN-γ) or M(IL-4) phenotype using human IFN-γ or IL-4, respectively. Parts of M1 or M2 macrophages were reprogrammed with IL-4 or IFN-γ, respectively, to induce the phenotypic switch. The representative markers of macrophage activation were analyzed by real-time PCR. Data were expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Skewing macrophages toward M(IL-4) phenotype confers human hepatocytes enhanced apoptosis resistance
We also assessed the effects of phenotypic switch of polarized human PBMC-derived macrophages on hepatocyte apoptosis. HepG2 and HL-7702 cells were pre-treated with CMs from M0, M(IFN-γ), M(IFN-γ→IL-4), M(IL-4), and M(IL-4→IFN-γ) macrophages, then hepatocyte apoptosis was induced by human TNF-α/
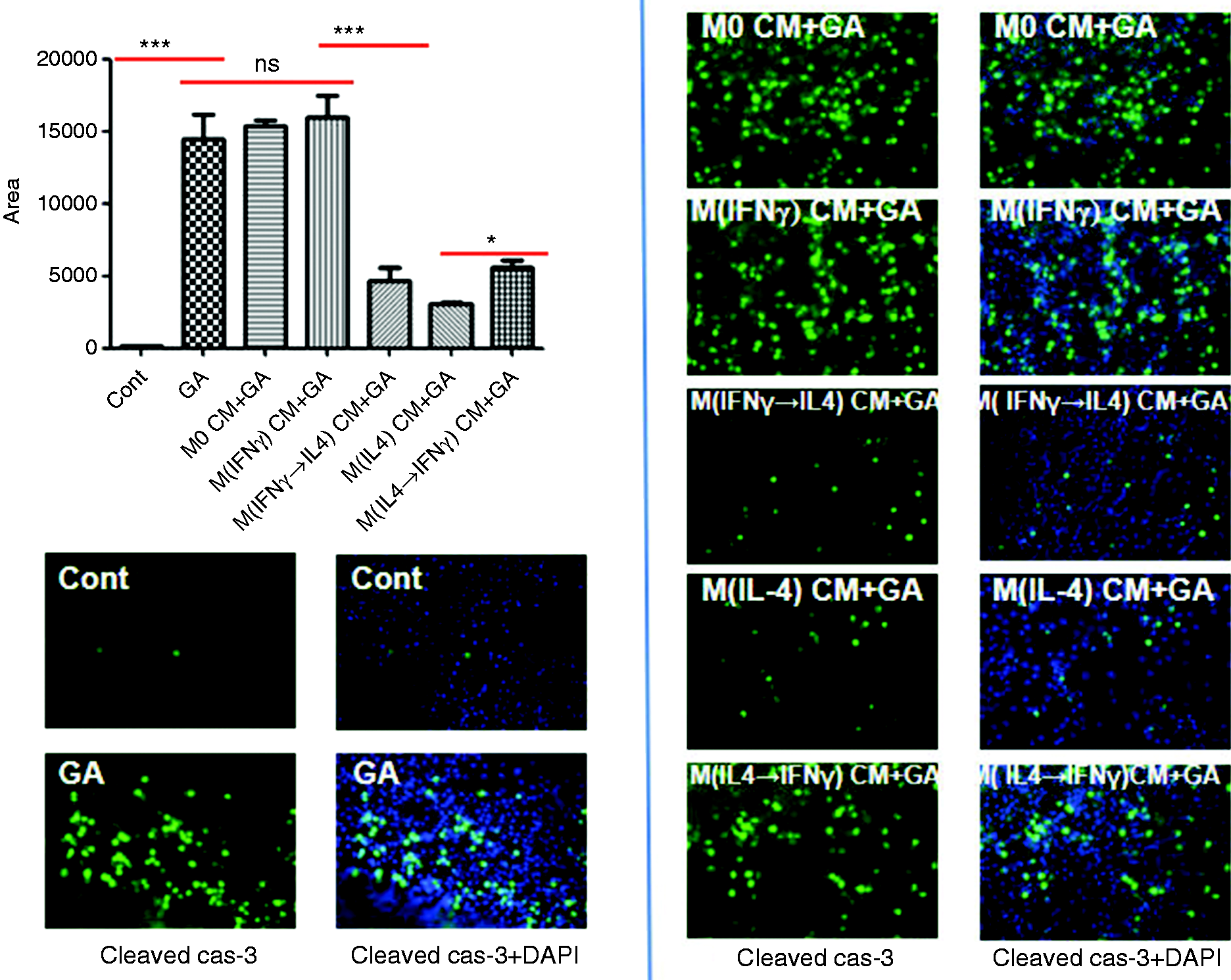
Skewing macrophages toward M(IL-4) phenotype confers HepG2 cells enhanced apoptosis resistance. Conditioned media from human M0, M(IFN-γ), M(IFN-γ→IL-4), M(IL-4), and M(IL-4→IFN-γ) macrophages were applied to HepG2 cells for 6 h. Cell apoptosis was induced by human TNF-α (50 µg/mL, Peprotech)/
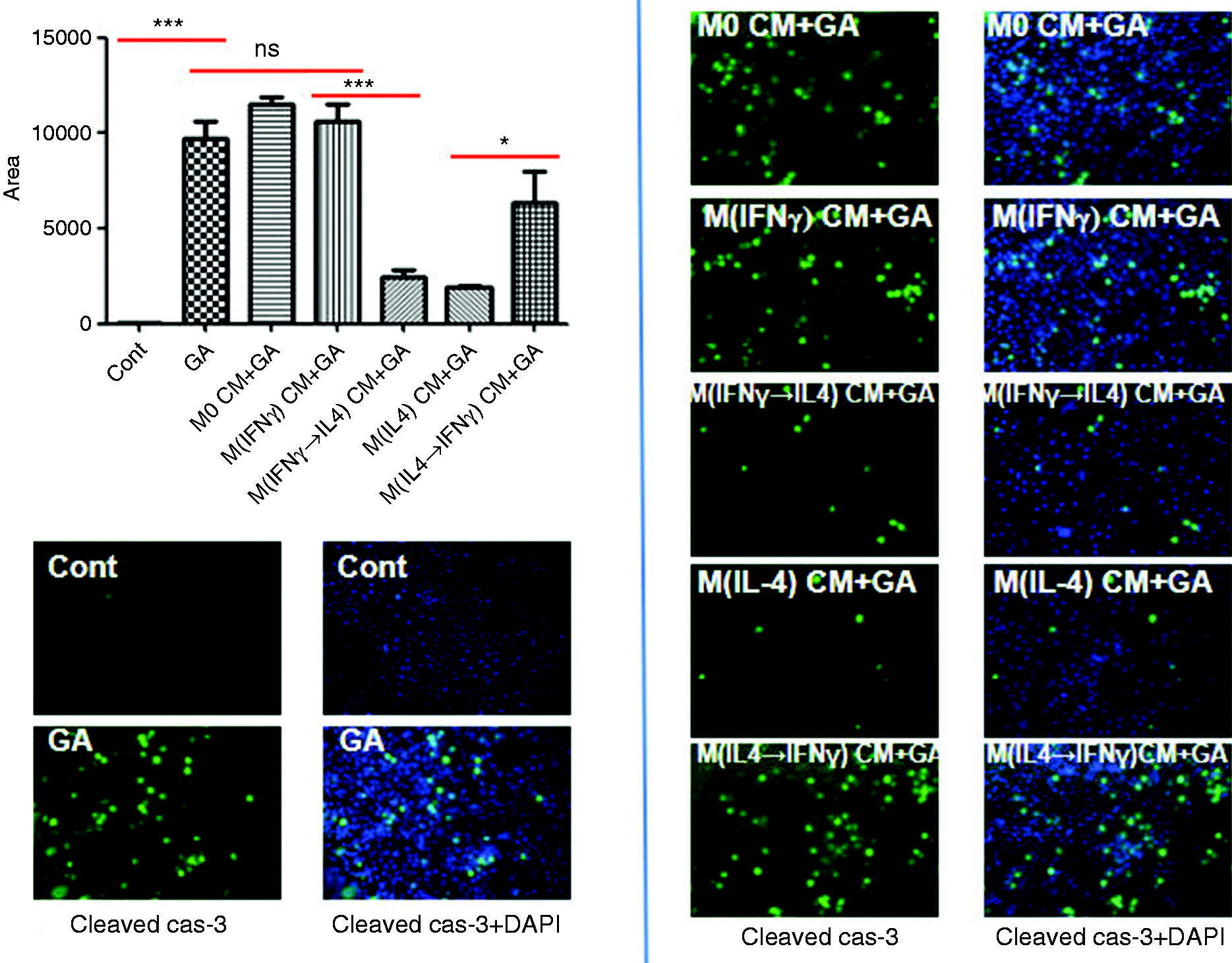
Skewing macrophages toward M(IL-4) phenotype confers HL-7702 cells enhanced apoptosis resistance. Conditioned media from human M0, M(IFN-γ), M(IFN-γ→IL-4), M(IL-4), and M(IL-4→IFN-γ) macrophages were applied to HL-7702 cells for 6 h. Cell apoptosis was induced by human TNF-α (50 µg/mL, Peprotech)/
The reprogramming of macrophages from ACLF patients
Furthermore, we analyzed the plasticity of macrophages derived from PBMCs of ACLF patients. As expected, IFN-γ treatment triggered the phenotypic switch of macrophages toward an M(IFN-γ) activation, as evidenced by high expression of M(IFN-γ) markers including CD86, TNF-α, iNOS, CD64, and CXCL10. On the other hand, IL-4 stimulus prompted the phenotypic switch of macrophages toward an M(IL-4) activation, as shown by high mRNA levels of M(IL-4) markers including IL-10, TGF-β, and IL-13 (Figure 7). Thereby, macrophages from ACLF patients can also be reprogrammed in response to diverse stimuli.
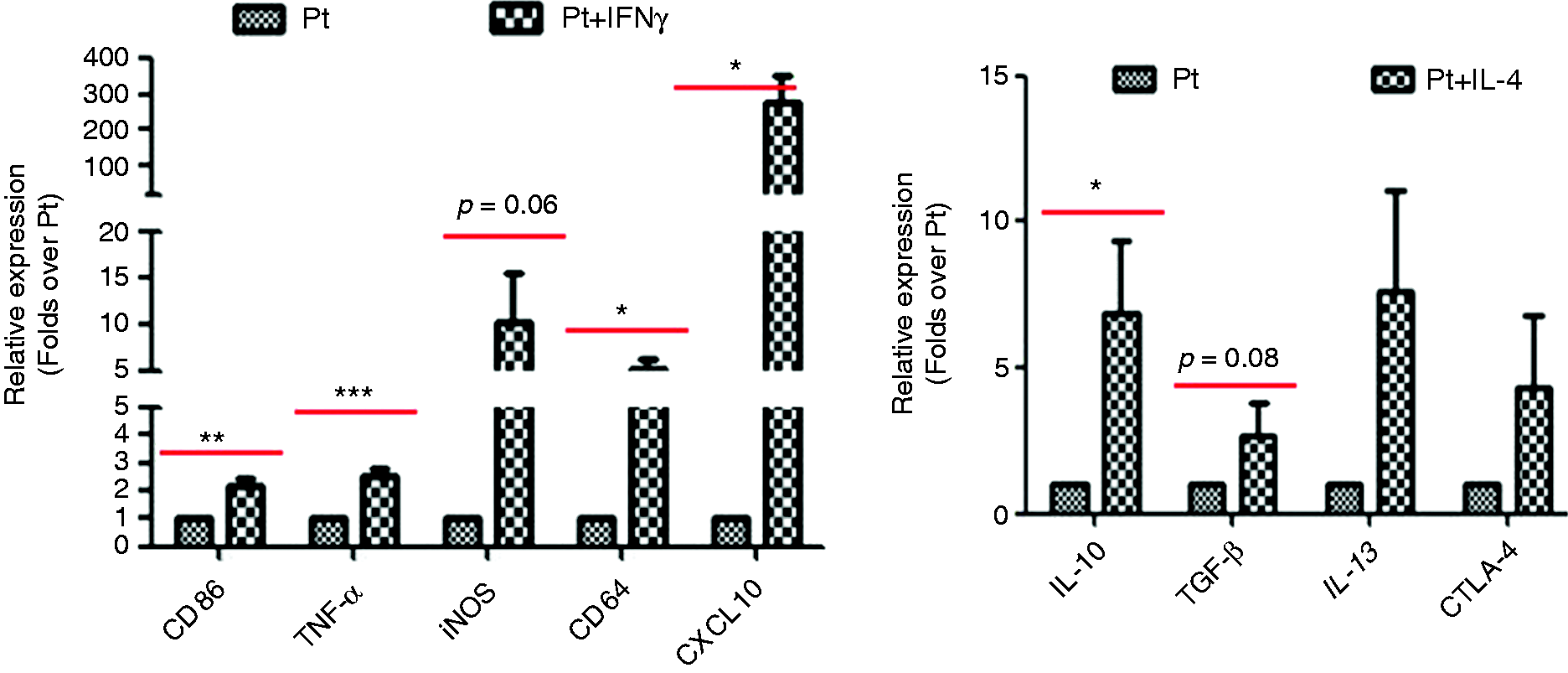
The phenotypic switch of PBMC-derived macrophages from ACLF patients in vitro. PBMC-derived macrophages from ACLF patients were polarized into M(IFN-γ) or M(IL-4) phenotype using human IFN-γ or IL-4, respectively. Parts of M(IFN-γ) or M(IL-4) macrophages were reprogrammed with IL-4 or IFN-γ, respectively, to induce the phenotypic switch. The representative markers of macrophage activation were analyzed by real-time PCR. Data were expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
ACLF is an increasingly recognized entity which carries a significant burden on critical care services and health care resources. However, the exact pathogenesis of ACLF remains to be elucidated, and novel treatments are desperately required1. In recent years, we have endeavored to investigate the pathogenesis of ACLF, and focus on injury resistance in the setting of hepatic fibrosis (simulating the development of ACLF). We previously have documented the favorable protection against apoptosis conferred by M2-like macrophages in mice (in vivo and in vitro) and humans (in vitro).10,14 In the present work, we highlight the phenotypic switch of human and mouse macrophages and the effect of this switch on hepatocyte apoptosis. We show that macrophages derived from PBMCs of healthy donors and murine liver, even PBMCs of patients with ACLF, can be re-programmed and shape their phenotype in response to diverse microenvironmental cues. Most importantly, we provide powerful evidence that skewing macrophages toward an M(IL-4) phenotype confers hepatocytes enhanced resistance to apoptosis in human and mice. In view of the dynamics and complexity of macrophage activation, study on phenotypic switch of macrophages, especially the phenotypic switch occurring in macrophages derived from ACLF patients, is required and clinically relevant.
ACLF is an innate immune-driven disorder, in which monocytes/macrophages are key determinants of the initiation, propagation, and resolution of liver injury.29–31 Macrophages inherently display tissue and environment-dependent plasticity, which means the same macrophages may have a variety of functions depending on the local tissue environment during different stages of disease.32,33 Macrophages are coarsely classified into two subsets: M1 macrophages and M2 macrophages. M1 macrophages are activated by pathogens or toxins (such as LPS), and secrete pro-inflammatory mediators which induces inflammation and liver damage; conversely, M2 macrophages are activated by IL-4/IL-13, and release anti-inflammatory or pro-resolving mediators which mediates wound repair, tissue remodeling and fibrosis.16,34 Macrophages in vivo adopt a mixed phenotype between M1- and M2-type macrophages. The M1/M2 balance is regarded as a decisive factor for macrophage function.31,35 Nevertheless, the classification of “M1” or “M2” macrophages is too simplistic to reflect the complexity of macrophage activation. Therefore, in this in vitro study, we nominate macrophages in response to IFN-γ stimulation as M(IFN-γ) and those upon IL-4 stimulation as M(IL-4), according to the nomenclature for macrophage activation proposed by Murray et al.36
We previously have documented the favorable protection conferred by M2-like macrophages in mice and human. In view of the great plasticity of macrophages, we extend our work from macrophage activation to their phenotypic switch. In vivo and in vitro studies have demonstrated that macrophages can undergo dynamic transitions, which is called polarization skewing or reprogramming.16,17,37 In keeping with this finding, our data show that the activation phenotype of M(IFN-γ) or M(IL-4) macrophages is skewed into M (IFN-γ→IL-4) or M(IL-4→IFN-γ) phenotype in response to IL-4 or IFN-γ stimulus, respectively. Importantly, this work focuses on the effects of macrophage reprogramming on the apoptosis resistance in human and mouse hepatocytes. According to our results, the CMs from M(IFN-γ→IL-4) macrophages confer human and mouse hepatocytes enhanced resistance to apoptosis induced by TNF-α/
In sum, our data further demonstrate the favorable protection of M(IL-4) macrophages from the viewpoint of phenotypic switch. Although our data are preliminary, it still provides evidence, at least in part, for the immune-modulating therapy of ACLF patients through re-orientating macrophages to acquire the desirable functions.
Footnotes
Declaration of conflicting interests
The author(s) declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Science and Technology Key Project (grant numbers 2017ZX10201201, 2017ZX10203201-005, 2017ZX10202203-006-001 and 2017ZX10302201-004-002), National Key R&D Program of China (grant number 2017YFA0103000), Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (grant number ZYLX201806), the Beijing Municipal Administration of Hospital’s Ascent Plan (grant number DFL20151601), the Digestive Medical Coordinated Development Center of Beijing Municipal Administration of Hospitals (grant number XXZ0503), the Beijing Municipal Science & Technology Projects (grant numbers Z151100004015066 and Z171100002217070), the YouAn fund for liver diseases and AIDS (grant numbers YNKT20160012 and YNKTTS201801189), and the Basic-Clinical Cooperation Project of Capital Medical University (grant number 17JL47).